
Notes:
- For details on how to use the published version v1.0.1 of workflows for scRNA-Seq data analysis in SciDAP refer to the Tutorials page.
- For up to date workflow description see Wiki page.
- Although, we eager to make our pipelines as reproducible as possible, certain issues with Seurat may affect the reproducibility even for containerized tools (see Reproducibility issue #5358)
Publications:
- Aizhan Surumbayeva, Michael Kotliar, Linara Gabitova-Cornell, Andrey Kartashov, Suraj Peri, Nathan Salomonis, Artem Barski, Igor Astsaturov, Preparation of mouse pancreatic tumor for single-cell RNA sequencing and analysis of the data, STAR Protocols, Volume 2, Issue 4, 2021, 100989, ISSN 2666-1667, https://doi.org/10.1016/j.xpro.2021.100989
- Kotliar M, Kartashov A and Barski A. CWL toolkit for single-cell sequencing data analysis [version 1; not peer reviewed]. F1000Research 2022, 11:819 (poster) (https://doi.org/10.7490/f1000research.1119046.1)
Minimum software requirements:
- cwltool or alternative CWL runner supporting v1.0
- Docker or Singularity container runtime environment
How to use it
This repository contains R scripts, CWL tools and examples of CWL workflows for single-cell RNA-Seq and Multiome data analyses.
Each R script can be run directly from the command line following the --help message instructions. However, to guarantee results reproducibility we containerized them and wrapped in CWL format.
CWL tools can be combined into the workflows depending on the type of input datasets and required complexity of the analysis. For example, for single-cell RNA-Seq use 1(a) – 2(a) – 3(a) and optionally 4(a) – 5(a,b); for Multiome ATAC-Seq and RNA-Seq use 1(b) – 2(b) – 2(a) – 3(a) - 3(b) - 3(c) and optionally 4(a) – 5(a,b).
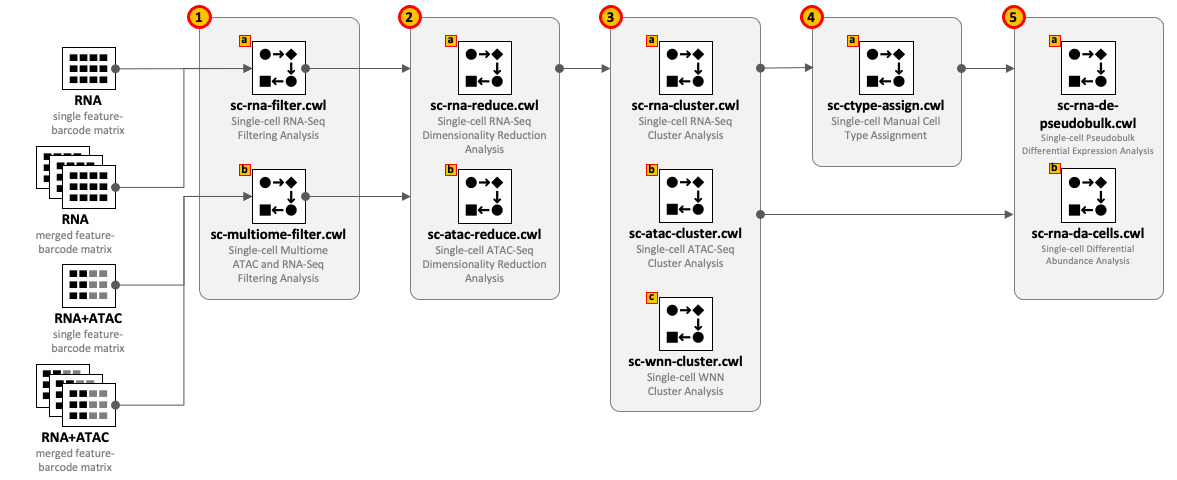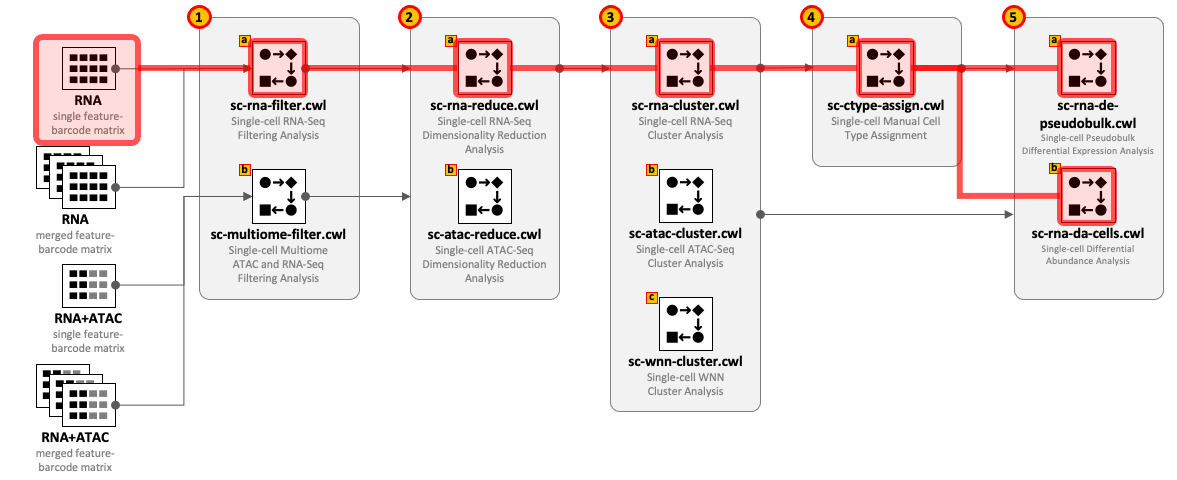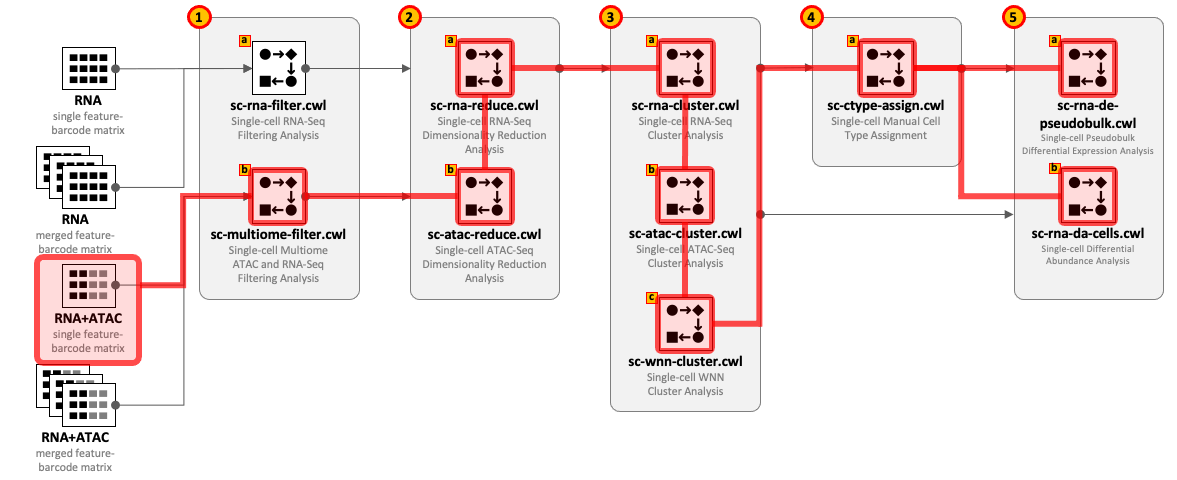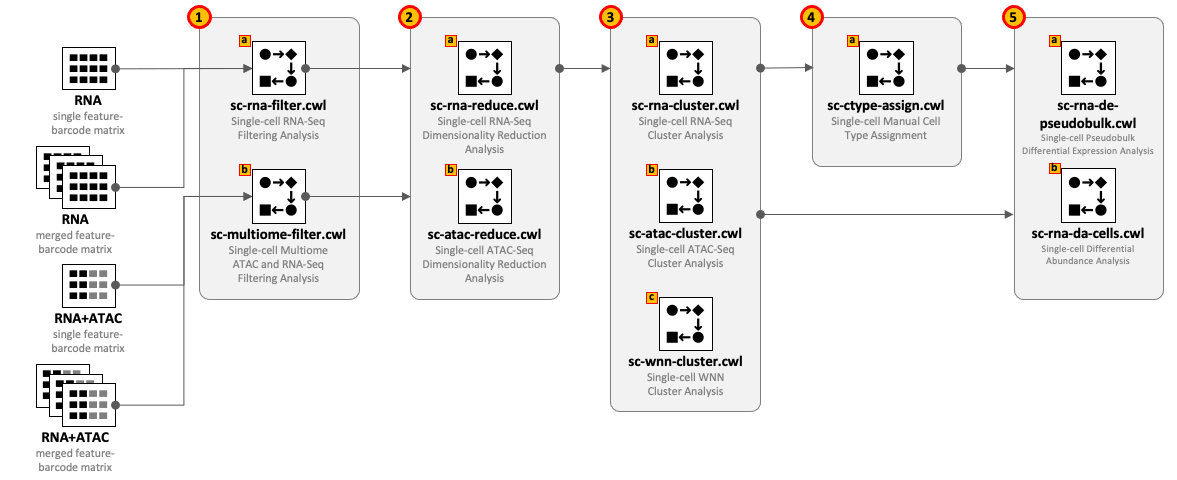
All CWL tools are divided into groups to cover the major steps of data analysis. For integrity reasons we recommend starting from the raw FASTQ files and use one of the Cell Ranger based pipelines from the Data preprocessing group. The results of these pipelines can be optionally exported into UCSC Cell Browser (see Visualization group).
Both sc-rna-filter.cwl and sc-multiome-filter.cwl tools use feature-barcode matrices as the main inputs. All other tools from the scRNA-Seq, scATAC-Seq and Multiome, and Secondary analyses groups exchange data through RDS files.
Data preprocessing
| Name | Description |
|---|---|
| cellranger-mkref.cwl | Builds Cell Ranger compatible reference folder from the custom genome FASTA and gene GTF annotation files |
| cellranger-count.cwl | Quantifies gene expression from a single-cell RNA-Seq library |
| cellranger-aggr.cwl | Aggregates outputs from multiple runs of Cell Ranger Count Gene Expression |
| cellranger-arc-mkref.cwl | Builds Cell Ranger ARC compatible reference folder from the custom genome FASTA and gene GTF annotation files |
| cellranger-arc-count.cwl | Quantifies chromatin accessibility and gene expression from a single-cell Multiome ATAC/RNA-Seq library |
| cellranger-arc-aggr.cwl | Aggregates outputs from multiple runs of Cell Ranger ARC Count Chromatin Accessibility and Gene Expression |
Visualization
| Name | Description |
|---|---|
| cellbrowser-build-cellranger.cwl | Exports clustering results from Cell Ranger Count Gene Expression and Cell Ranger Aggregate experiments into compatible with UCSC Cell Browser format |
| cellbrowser-build-cellranger-arc.cwl | Exports clustering results from Cell Ranger ARC Count Chromatin Accessibility and Gene Expression or Cell Ranger ARC Aggregate experiments into compatible with UCSC Cell Browser format |
scRNA-Seq
| Name | Description |
|---|---|
| sc-rna-filter.cwl | Filters single-cell RNA-Seq datasets based on the common QC metrics |
| sc-rna-reduce.cwl | Integrates multiple single-cell RNA-Seq datasets, reduces dimensionality using PCA |
| sc-rna-cluster.cwl | Clusters single-cell RNA-Seq datasets, identifies gene markers |
scATAC-Seq and Multiome
| Name | Description |
|---|---|
| sc-multiome-filter.cwl | Filters single-cell multiome ATAC-Seq and RNA-Seq datasets based on the common QC metrics |
| sc-atac-reduce.cwl | Integrates multiple single-cell ATAC-Seq datasets, reduces dimensionality using LSI |
| sc-atac-cluster.cwl | Clusters single-cell ATAC-Seq datasets, identifies differentially accessible peaks |
| sc-wnn-cluster.cwl | Clusters multiome ATAC-Seq and RNA-Seq datasets, identifies gene markers and differentially accessible peaks |
Secondary analyses
| Name | Description |
|---|---|
| sc-ctype-assign.cwl | Assigns cell types for clusters based on the provided metadata file |
| sc-rna-de-pseudobulk.cwl | Identifies differentially expressed genes between groups of cells coerced to pseudobulk datasets |
| sc-rna-da-cells.cwl | Detects cell subpopulations with differential abundance between datasets split by biological condition |
| sc-triangulate.cwl | Harmonizes conflicting annotations in single-cell genomics studies using scTriangulate |
Utilities
| Name | Description |
|---|---|
| tar-extract.cwl | Extracts the content of TAR file into a folder |
| tar-compress.cwl | Creates compressed TAR file from a folder |
Workflow examples for scRNA-Seq analysis
| Name | Description |
|---|---|
| sc-ref-indices-wf.cwl | Builds a Cell Ranger and Cell Ranger ARC compatible reference folders from the custom genome FASTA and gene GTF annotation files |
| sc-rna-align-wf.cwl | Runs Cell Ranger Count to quantify gene expression from a single-cell RNA-Seq library |
| sc-rna-aggregate-wf.cwl | Aggregates gene expression data from multiple Single-cell RNA-Seq Alignment experiments |
| sc-rna-analyze-wf.cwl | Runs filtering, normalization, scaling, integration (optionally) and clustering for a single or aggregated single-cell RNA-Seq datasets |
Workflow examples for Multiome analysis
| Name | Description |
|---|---|
| sc-multiome-align-wf.cwl | Runs Cell Ranger ARC Count to quantifies chromatin accessibility and gene expression from a single-cell Multiome ATAC and RNA-Seq library |
| sc-multiome-aggregate-wf.cwl | Aggregates data from multiple Single-cell Multiome ATAC and RNA-Seq Alignment experiments |
| sc-multiome-analyze-wf.cwl | Runs filtering, normalization, scaling, integration (optionally) and clustering for a single or aggregated single-cell Multiome ATAC-Seq and RNA-Seq datasets |





