Comments (6)
 davhum
commented on May 28, 2024
davhum
commented on May 28, 2024
Hi Nelson,
Thanks for trying out our new package. We suspect the issue you are experiencing is due to the passing of a fasta file as a genome object. i.e. this line
genome <- "/media/ot/Data/Nelson/refdata-cellranger-GRCh38-3.0.0/fasta"
Instead you should set the genome object to a BSgenome S4 data object from the appropriate BSgenome package. i.e.
genome <- BSgenome.Hsapiens.UCSC.hg38::BSgenome.Hsapiens.UCSC.hg38
If you don't already have this package you can download it via the following commands:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("BSgenome.Hsapiens.UCSC.hg38")
Let us know if this fixes the problem.
Regards,
Dave
from sierra.
 zezhuo
commented on May 28, 2024
zezhuo
commented on May 28, 2024
Hi Dave,
Thanks for your prompt reply. It solved the problem.
But when I move on to SplitBam function, No data found. I have tested several genes. Bam file is from the output of CellRanger. What is the problem with my data set?
Regards,
Nelson
SplitBam function:
> str(cells.df)
'data.frame': 1872 obs. of 2 variables:
$ celltype: Factor w/ 4 levels "cartilaginous",..: 1 3 3 3 3 3 1 3 3 2 ...
$ cellbc : chr "AAACCTGGTTAAGGGC" "AAACCTGTCGAGAGCA" "AAACGGGAGAACAATC" "AAACGGGAGAATCTCC" ...
>
> SplitBam(bam = "./L07/possorted_genome_bam.bam",
+ cellbc.df = cells.df,
+ outdir = outdir,
+ gtf_gr = gtf_gr,
+ geneSymbol = "ENG")
splitting bam file: ./L07/possorted_genome_bam.bam
[1] "cartilaginous" "stress" "angiogenic" "fibroblast"
processing cell type cartilaginous
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
No data found for cartilaginous
processing cell type stress
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
No data found for stress
processing cell type angiogenic
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
No data found for angiogenic
processing cell type fibroblast
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
No data found for fibroblast
Additional scripts:
### Visualization
library(Seurat)
library(stringr)
library(dplyr)
#Previous definitions
out.dir <- "example_TIP_aggregate"
#Read in the counts
peak.counts <- ReadPeakCounts(data.dir = out.dir)
# Correct cell id
colnames(peak.counts) <- colnames(peak.counts) %>% str_split("-") %>% sapply("[[", 1)
#Read in peak annotations
peak.annotations <- read.table("TIP_merged_peak_annotations.txt",
header = TRUE,
sep = "\t",
row.names = 1,
stringsAsFactors = FALSE)
head(peak.annotations)
#Load precompiled gene-level object called 'genes.seurat'
Low_L07 <- readRDS("../scRNAseq/Low_L07.rds")
peaks.seurat <- PeakSeuratFromTransfer(peak.data = peak.counts,
genes.seurat = Low_L07,
annot.info = peak.annotations,
min.cells = 0, min.peaks = 0)
# Testing for differential transcript usage
res.table = DUTest(peaks.seurat,
population.1 = "stress",
population.2 = "fibroblast",
exp.thresh = 0.1,
feature.type = c("UTR3", "exon"))
res.table.top <- subset(res.table, abs(Log2_fold_change) > 1)
head(res.table.top)
# Plotting gene expression using the Seurat FeaturePlot function
Seurat::FeaturePlot(Low_L07, "ENG", cols = c("lightgrey", "red"))
# Select top peaks DU within the MMP3 gene
peaks.to.plot <- rownames(subset(res.table.top, gene_name == "ENG"))
PlotRelativeExpressionUMAP(peaks.seurat, peaks.to.plot = peaks.to.plot)
### Sierra coverage plots
cells.df <- Idents(Low_L07) %>% as.data.frame()
cells.df$cellbc <- rownames(cells.df)
colnames(cells.df) <- c("celltype", "cellbc")
gtf_gr <- rtracklayer::import("genes.gtf")
outdir = "bam_subsets/"
dir.create(outdir)
SplitBam(bam = "./L07/possorted_genome_bam.bam",
cellbc.df = cells.df,
outdir = outdir,
gtf_gr = gtf_gr,
geneSymbol = "ENG")
from sierra.
 rj-patrick
commented on May 28, 2024
rj-patrick
commented on May 28, 2024
Hi Nelson,
If you're using CellRanger, it's possible you need a '-1' character appended to your cell barcodes.
Try the following code, and let me know if that helps:
cells.df$cellbc <- paste0(cells.df$cellbc, "-1")
Cheers,
- Ralph
from sierra.
zezhuo
commented on May 28, 2024
Hi Ralph,
Thanks for your suggestion, but still "No data found". Any other solution? Thanks!
Regards,
Nelson
> cells.df$cellbc <- paste0(cells.df$cellbc, "-1")
> str(cells.df)
'data.frame': 1872 obs. of 2 variables:
$ celltype: Factor w/ 4 levels "cartilaginous",..: 1 3 3 3 3 3 1 3 3 2 ...
$ cellbc : chr "AAACCTGGTTAAGGGC-1" "AAACCTGTCGAGAGCA-1" "AAACGGGAGAACAATC-1" "AAACGGGAGAATCTCC-1" ...
> SplitBam(bam = "./L07/possorted_genome_bam.bam",
+ cellbc.df = cells.df,
+ outdir = outdir,
+ gtf_gr = gtf_gr,
+ geneSymbol = "ENG")
splitting bam file: ./L07/possorted_genome_bam.bam
[1] cartilaginous stress angiogenic fibroblast
Levels: cartilaginous angiogenic stress fibroblast
processing cell type cartilaginous
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
Writing to bam_subsets/cartilaginous.ENG.bam
processing cell type stress
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
Writing to bam_subsets/stress.ENG.bam
processing cell type angiogenic
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
Writing to bam_subsets/angiogenic.ENG.bam
processing cell type fibroblast
[W::hts_idx_load2] The index file is older than the data file: ./L07/possorted_genome_bam.bam.bai
chunk0: 8192 length of aln: 0
Writing to bam_subsets/fibroblast.ENG.bam
> sessionInfo()
R version 3.6.1 (2019-07-05)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.3 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.7.1
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.7.1
locale:
[1] LC_CTYPE=en_HK.UTF-8 LC_NUMERIC=C LC_TIME=en_HK.UTF-8 LC_COLLATE=en_HK.UTF-8 LC_MONETARY=en_HK.UTF-8
[6] LC_MESSAGES=en_HK.UTF-8 LC_PAPER=en_HK.UTF-8 LC_NAME=C LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_HK.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] dplyr_0.8.3 stringr_1.4.0 Seurat_3.1.1 Sierra_0.1.0
loaded via a namespace (and not attached):
[1] reticulate_1.13 R.utils_2.9.0 tidyselect_0.2.5 RSQLite_2.1.3
[5] AnnotationDbi_1.48.0 htmlwidgets_1.5.1 grid_3.6.1 BiocParallel_1.20.0
[9] Rtsne_0.15 munsell_0.5.0 codetools_0.2-16 ica_1.0-2
[13] statmod_1.4.32 future_1.15.1 colorspace_1.4-1 Biobase_2.46.0
[17] knitr_1.26 rstudioapi_0.10 stats4_3.6.1 SingleCellExperiment_1.8.0
[21] ROCR_1.0-7 gbRd_0.4-11 listenv_0.7.0 labeling_0.3
[25] Rdpack_0.11-0 GenomeInfoDbData_1.2.2 hwriter_1.3.2 bit64_0.9-7
[29] farver_2.0.1 vctrs_0.2.0 xfun_0.11 biovizBase_1.34.0
[33] BiocFileCache_1.10.2 R6_2.4.1 GenomeInfoDb_1.22.0 rsvd_1.0.2
[37] locfit_1.5-9.1 AnnotationFilter_1.10.0 bitops_1.0-6 DelayedArray_0.12.0
[41] assertthat_0.2.1 SDMTools_1.1-221.2 scales_1.1.0 nnet_7.3-12
[45] gtable_0.3.0 npsurv_0.4-0 globals_0.12.4 ensembldb_2.10.2
[49] rlang_0.4.2 zeallot_0.1.0 genefilter_1.68.0 splines_3.6.1
[53] rtracklayer_1.46.0 lazyeval_0.2.2 acepack_1.4.1 dichromat_2.0-0
[57] checkmate_1.9.4 reshape2_1.4.3 GenomicFeatures_1.38.0 backports_1.1.5
[61] Hmisc_4.3-0 tools_3.6.1 ggplot2_3.2.1 gplots_3.0.1.1
[65] RColorBrewer_1.1-2 BiocGenerics_0.32.0 ggridges_0.5.1 Rcpp_1.0.3
[69] plyr_1.8.4 base64enc_0.1-3 progress_1.2.2 zlibbioc_1.32.0
[73] purrr_0.3.3 RCurl_1.95-4.12 prettyunits_1.0.2 rpart_4.1-15
[77] openssl_1.4.1 pbapply_1.4-2 cowplot_1.0.0 S4Vectors_0.24.1
[81] zoo_1.8-6 SummarizedExperiment_1.16.0 ggrepel_0.8.1 cluster_2.1.0
[85] magrittr_1.5 data.table_1.12.6 lmtest_0.9-37 RANN_2.6.1
[89] ProtGenerics_1.18.0 fitdistrplus_1.0-14 matrixStats_0.55.0 hms_0.5.2
[93] lsei_1.2-0 xtable_1.8-4 XML_3.98-1.20 IRanges_2.20.1
[97] gridExtra_2.3 compiler_3.6.1 biomaRt_2.42.0 tibble_2.1.3
[101] KernSmooth_2.23-16 crayon_1.3.4 R.oo_1.23.0 htmltools_0.4.0
[105] Formula_1.2-3 tidyr_1.0.0 geneplotter_1.64.0 RcppParallel_4.4.4
[109] DBI_1.0.0 dbplyr_1.4.2 MASS_7.3-51.4 rappdirs_0.3.1
[113] Matrix_1.2-18 R.methodsS3_1.7.1 gdata_2.18.0 parallel_3.6.1
[117] Gviz_1.30.0 metap_1.1 igraph_1.2.4.2 GenomicRanges_1.38.0
[121] pkgconfig_2.0.3 GenomicAlignments_1.22.1 foreign_0.8-72 plotly_4.9.1
[125] foreach_1.4.7 annotate_1.64.0 XVector_0.26.0 bibtex_0.4.2
[129] DEXSeq_1.32.0 VariantAnnotation_1.32.0 BSgenome.Hsapiens.UCSC.hg38_1.4.1 digest_0.6.23
[133] sctransform_0.2.0 RcppAnnoy_0.0.14 tsne_0.1-3 Biostrings_2.54.0
[137] leiden_0.3.1 htmlTable_1.13.2 uwot_0.1.4 curl_4.3
[141] Rsamtools_2.2.1 gtools_3.8.1 lifecycle_0.1.0 nlme_3.1-142
[145] jsonlite_1.6 viridisLite_0.3.0 askpass_1.1 BSgenome_1.54.0
[149] pillar_1.4.2 lattice_0.20-38 httr_1.4.1 survival_2.44-1.1
[153] glue_1.3.1 png_0.1-7 iterators_1.0.12 bit_1.1-14
[157] stringi_1.4.3 blob_1.2.0 DESeq2_1.26.0 latticeExtra_0.6-28
[161] caTools_1.17.1.3 memoise_1.1.0 irlba_2.3.3 future.apply_1.3.0
[165] ape_5.3
from sierra.
rj-patrick
commented on May 28, 2024
Hi Nelson,
I don't see the 'no data found' message in your output after the cell barcodes were updated - the messages indicate the BAM files should have been written. Can you clarify if the BAM files have not been produced in your output directory?
Cheers,
- Ralph
from sierra.
zezhuo
commented on May 28, 2024
Hi Ralph,
Sorry. I check the output dir and get the bam files. It did solve the problem.
Thanks you so much for your kindly help. I love the coverage plot.
Regards,
Nelson
bam.files <- paste0(outdir, dir(outdir, "ENG.bam$"))
PlotCoverage(genome_gr = gtf_gr,
geneSymbol = "ENG",
genome = "mm10",
bamfiles = bam.files)
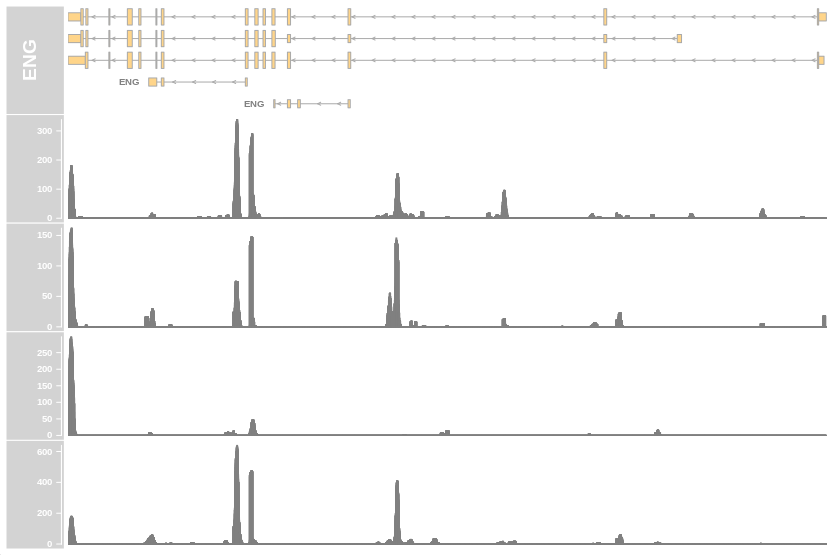
from sierra.
Related Issues (20)
- Using data with batch effects in Sierra HOT 2
- Error in peak calling HOT 2
- peak discrepancy HOT 1
- Error when running CountPeaks HOT 8
- CoveragePlot error with 'zoom_3UTR=TRUE ' HOT 1
- Which alignment method and indexing options are suitable to use with Sierra? HOT 3
- Does the Sierra package have a detailed protocol, I would like to find. HOT 1
- Chromosome name in FindPeaks // Help with Output HOT 3
- Generate GitHub Releases HOT 2
- Cellranger mkref function parameters for Sierra HOT 1
- FindPeaks Error--'x' values larger than vector length 'sum(width)' HOT 2
- DUTest function Error HOT 1
- Sierra dataframe has 0 length HOT 1
- [E::hts_open_format] Failed to open file HOT 1
- is it possible to generate a plot that shows global 3'UTR length change?
- issues with generating splice junction file HOT 7
- MergePeakCoordinates takes long time! HOT 9
- Paired-end & PlotRelativeExpression functions. HOT 1
- Getting "Error in (function (x) : attempt to apply non-function" HOT 3
- Using Sierra with Singleron Biotechnologies Platform HOT 1
Recommend Projects
-
 React
React
A declarative, efficient, and flexible JavaScript library for building user interfaces.
-
Vue.js
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
-
 Typescript
Typescript
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
-
TensorFlow
An Open Source Machine Learning Framework for Everyone
-
Django
The Web framework for perfectionists with deadlines.
-
 Laravel
Laravel
A PHP framework for web artisans
-
D3
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
-
Recommend Topics
-
javascript
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
-
web
Some thing interesting about web. New door for the world.
-
server
A server is a program made to process requests and deliver data to clients.
-
Machine learning
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
-
Visualization
Some thing interesting about visualization, use data art
-
Game
Some thing interesting about game, make everyone happy.
Recommend Org
-
Facebook
We are working to build community through open source technology. NB: members must have two-factor auth.
-
Microsoft
Open source projects and samples from Microsoft.
-
Google
Google ❤️ Open Source for everyone.
-
Alibaba
Alibaba Open Source for everyone
-
D3
Data-Driven Documents codes.
-
Tencent
China tencent open source team.
from sierra.