![]()
We introduce Signac, a versatile R package to facilitate the analysis workflow for single-cell data. It helps to find marker genes faster and more accurate, search for cells with similar expression profiles, integrate multiple datasets in the BioTuring Browser database (know more about BioTuring Browser), etc. For users with a limited computational resource, we provide the helper functions to exercise all analyses for the large-scale datasets from disk. Because of its speed and flexibility, it can be adapted to any existing R analysis pipeline to help explore single-cell data more efficient.
This package can also be used as the reference for the computational methods used in BioTuring Browser software.
Please visit this blog to read about Venice, a fast and accurate method for finding marker genes, which is incorporated into Signac. Venice function can be accessed via Signac::VeniceMarker
You can also read the manuscript about Venice here.
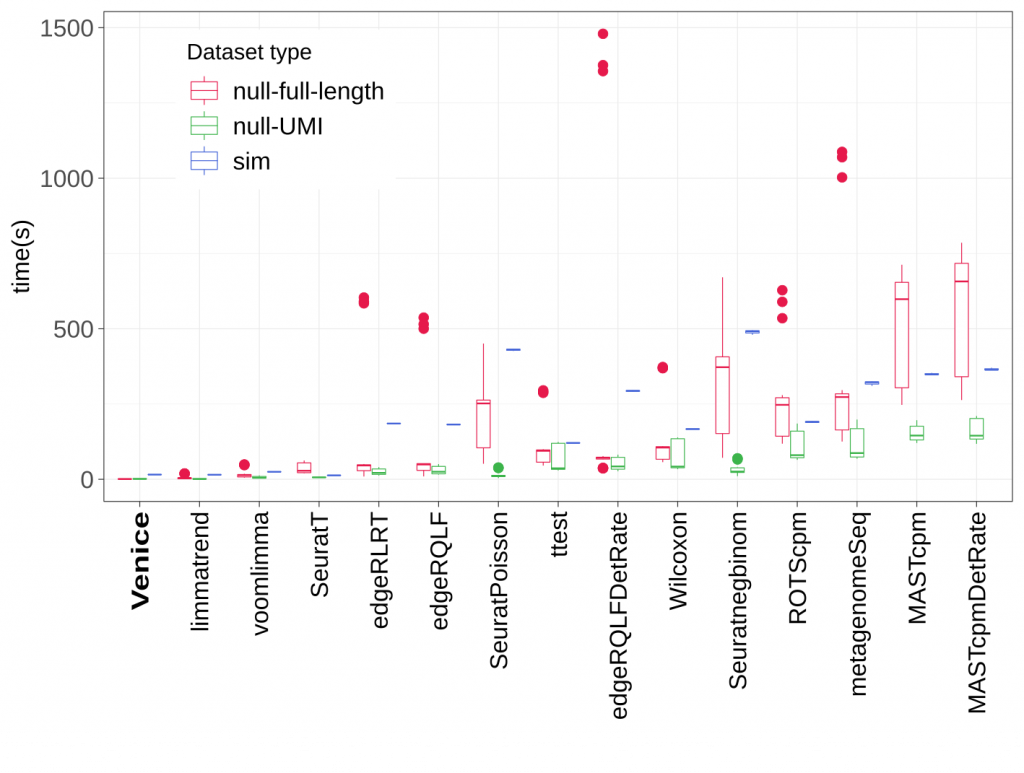
Below is the benchmark when finding 4 types of DE genes in a simulated dataset (using scDD R package). Total 15 methods included Venice are tested. The dataset has 2000 DE genes that are even divided into 4 groups (DE, DP, DB, DM), and 18000 non-DE genes (EP and EE).
devtools::install_github("bioturing/signac")### The pbmc object created by following the tutorial: https://satijalab.org/seurat/v3.1/pbmc3k_tutorial.html
> pbmc
An object of class Seurat
13714 features across 2638 samples within 1 assay
Active assay: RNA (13714 features, 2000 variable features)
1 dimensional reduction calculated: pca
> head(Seurat::Idents(pbmc))
AAACATACAACCAC-1 AAACATTGAGCTAC-1 AAACATTGATCAGC-1 AAACCGTGCTTCCG-1
1 3 1 2
AAACCGTGTATGCG-1 AAACGCACTGGTAC-1
6 1
Levels: 0 1 2 3 4 5 6 7 8
> ### Find markers for NK cells (cluster 6)
> VeniceMarker(pbmc@assays$RNA@counts, cluster=Idents(pbmc) == 6) %>% head
Gene.ID Gene.Name Dissimilarity Bin.count Log10.p.value Perm.p.value
1 1804 GNLY 0.8129016 3 -102.95623 NaN
2 13002 NKG7 0.8186809 4 -102.32274 NaN
3 9294 GZMB 0.8004017 3 -101.14819 NaN
4 7153 PRF1 0.7359742 3 -93.08173 NaN
5 11880 CST7 0.6679871 3 -84.52626 NaN
6 3202 FGFBP2 0.6670391 3 -84.37497 NaN
Log10.adjusted.p.value Up.Down.score Log2.fold.change pct1 pct2
1 -98.81906 1 4.016472 96.12903 13.129279
2 -98.48661 1 3.424045 100.00000 25.493355
3 -97.48815 1 3.174406 96.12903 6.806283
4 -89.54662 1 2.502070 94.83871 10.672573
5 -81.08807 1 2.038486 94.83871 14.941603
6 -81.01596 1 2.362991 87.74194 6.202175























