paper's People
Contributors

Stargazers






Watchers
paper's Issues
[2022] Xtal2DoS: Attention-based Crystal to Sequence Learning for Density of States Prediction
概要
DoSなどの系列データを予測するXtal2DoSを提案。エッジをattentionに加えたGATで潜在特徴を生成し、transformer型のアーキテクチャで系列データを出力する。DoS, phonon-DoSデータの予測でSoTA更新。
論文リンク
https://openreview.net/forum?id=Fw8PO9i5KG
著者/所属機関
Junwen Bai and Yuanqi Du and Yingheng Wang and Shufeng Kong and John Gregoire and Carla P Gomes
投稿日付
2022/9/27
背景
結晶構造を対象にした機械学習はスカラー量の例が多く、DoS(phonon density of states: phDoS, electronic density of states: eDoS)などのスペクトルに関しての予測例が少ない。
単純にスペクトルを構成するスカラー量を一度に出力すると、連続的であるという要請を満たさない。Mat2Specはこの問題に対し対照学習、VAEを使用することで連続量の相関を扱っている。
新規性
transformer型のアーキテクチャを用いてDoSを予測するXtal2DoSを提案。
手法
モデル
ノードの特徴量としてCGCNNのembeddinbgを使用。これをGATで変換する。
attention部分は、ノード特徴量をq,k,vに変換する。このとき、kはエッジを変換したものも加えることでエッジの情報を取り込んだattention weightを出力する。

さらに残差接続を加え、元のノード特徴量との線形和をとることでノード特徴量の更新を行う。
最終層のノード特徴量の平均をとり、global featureとした。これを初期のmemory入力とし、以下の4種のモデルを検証した。
- RNN
- Chunk RNN
- Chunk RNN + Attention
- Transformer
RNNでは127個のスカラーデータを順番に出力し、DoSとする。Chunk RNNでは分割したDoSデータを順番に出力。これにattentionを加えたモデルも作成。

transformerのモデルではglobal featureにpositional encodingを結合したものにした。この特徴量系列に対しself-attentionを行い、エンコードする。デコードでは潜在空間の特徴量とinputの原子とのattentionを行う。

結果
全ての指標においてこれまでのモデルより高精度

また、transformer型のアーキテクチャにすることで、他のRNNベースのモデルよりも高精度を実現

計算に必要な時間も他のモデルに比べ圧倒的に短かった。RNNベースのモデルより速いのはもちろんのこと、RNNの機構が含まれないMat2Specより速い。これは内部で対照学習を行なっているからとのこと(詳細は元論文参照)。
参考文献
[2018] Thermodynamic limit for synthesis of metastable inorganic materials
概要
合成可能なe_above_hull最大値を算出する論文。前駆体が分解し新しい結合を形成、新しい結晶ができるという合成プロセスに着目し、アモルファス構造よりもエネルギーが高い構造は合成不可であるとする。高圧相などの例外を除き、代表的な41組成においてfalse-negativeは0件であった。

論文リンク
https://www.science.org/doi/10.1126/sciadv.aaq0148
著者/所属機関
Muratahan Aykol,1* Shyam S. Dwaraknath,1 Wenhao Sun,2 Kristin A. Persson1,3†
1Energy Technologies Area, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. 2Materials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. 3Department of Materials Science and Engineering, Uni- versity of California, Berkeley, Berkeley, CA 94720, USA.
*Present address: Toyota Research Institute, Los Altos, CA 94022, USA. †Corresponding author. Email: [email protected]
投稿日付
2018/4/20
背景
-
e_above_hullに対し適当な閾値を設定しても合成可能性の指標として機能しない。
-
既存の準安定相はe_above_hullが大きく異なり、0.2eV/atom程度のものもある
新規性
-
前駆体が分解し新しい結合を形成、新しい結晶ができるという合成プロセスそのものに着目。
-
全ての結晶はアモルファス構造に競合するため、最安定なアモルファス構造よりもエネルギーが低い必要がある。→アモルファスリミット
手法
- アモルファス相よりもエネルギーがより高い場合は、いかなる温度でも合成できないと仮定。エントロピーはアモルファス相の方が大きいため、温度を上げても結晶が安定になることはない(下図のA)。合成時には必ずアモルファス構造を経由するという過程のもと、安定なアモルファス相であれば結晶化はしないということ。

-
全てのアモルファス構造はサンプリングできないので、計算した中で最も安定なものをアモルファスリミットとした。より多くのアモルファス構造を計算するとこれが下がる方向にはいくが、false-negativeを判定するのには問題ない。
-
ランダムに原子を配置し、3000〜5000Kで5000ステップMDをする。5つほどサンプルしクエンチしてアモルファス構造を生成。
結果
-
よく調べられている41組成で精度を検証。
-
エネルギーのヒストグラムを取ると、低エネルギー領域でアモルファス相とICSDの結晶相の重複部分が大きかった。そのため系によってはアモルファス相は強い競合相として存在していることを示す。
-
合成可能な材料は以下の4つの例外を除いてアモルファスリミットより低エネルギーであった。
- ICSDに掲載されていない仮想材料
- ICSDに掲載されている仮想材料
- ICSDに掲載されている高圧相
- 磁性によりエネルギーが正確でないもの
つまり合成可能な700の多形ではfalse-negativeは0であった。
-
酸化物のアモルファスリミットは0.05-0.5くらい、ガラス材料はアモルファスリミットが小さい。
-
窒化物は他の系に比べてアモルファスリミットが高い。これは結合が固く柔軟性が無いので、アモルファスよりも結晶でいる方が安定になるため。
-
e_above_hullで安定性を評価するとき、閾値を0.1eV/atomとすると、閾値をを超える準安定相がたくさんあるが、アモルファスリミットだと取りこぼすことが無い。
-
アモルファスのサンプル数を増やすとアモルファスリミットは下がるが、多くは30 meV/atom程度。
参考文献
[2022] Chemistry Insights for Large Pretrained GNNs
概要
OC20を学習させたモデルが、化学的に正しい法則を学習できているかを調査。入力に変化を与える前後での予測値の変化を系統的に分析。吸着原子が系全体に与える影響が大きいこと、周期表で隣接した元素置換は、他の元素置換よりも差が小さくなること、力の大きさとエネルギー変化の間に正の相関があることなど、化学的直感に沿った結果が観測された。
論文リンク
https://openreview.net/forum?id=hNWJbH2lVW
著者/所属機関
Katherine Xu, Janice Lan
投稿日付
2022/11/5
背景
巨大なGNNモデルが構築されてきたが、これがなぜ、どのように機能しているかあまりわかっていない。これを理解することでモデルに生じる誤差の系統を決定することができ、予測が化学的に現実的なものかを判断することができる。
巨大なデータセット(OC20)で学習したモデルに対し、これまでの分析はモデルの重みを可視化するだけにとどまっていた。
化学分野でのGNNモデルの理解は画像や自然言語分野に比べてあまり行われてこなかった。
新規性
モデルを理解するため、対象に少しの変化を与え、前後でモデルの予測値がどのように変化したか分析する(perturbation analyses)。
手法
データセット
OC20のvalデータから10000個をランダムに取得。75個のユニークな吸着分子、4202個のユニークなバルク構造が得られ、OC20で用いられる全元素(56個)が含まれるデータセットを作成。元素一覧は以下

Input perturbation analysis
OC20のS2EFタスクを学習させたGemNEt-OCモデルを用いた。変化を与えた前後でエネルギーと力の予測値を比較する。単純化のため、一度に変化させる原子は1つに限定した。
変化は以下のパターンで行った。
- 原子を除去
- 原子位置を変更
- 原子番号を変更(周期表で2つ分の上下左右への移動に制限)
結果
それぞれの原子を除去したときのエネルギー変化を観測すると、吸着分子の原子を除去するのが最もエネルギー変化が大きく、バルク部分原子を除去するのが最も変化が小さかった。


次に、元素を変更したときのエネルギー変化を観測した。CとOに関して行った結果は以下。

2つ隣よりも1つ隣の方が変化が小さかった。
また、周期内での置換よりも族内での置換の方が変化が大きかったが、この結果は何が原因か現状は分からないとのこと。
また、より多くの元素で置換した結果、周期表で大きく離れた置換はエネルギー変化が大きい傾向であり、モデルは周期表で近い元素は特性も似ているということを学習できていることを示唆している。
次に、原子を、forceの方向に移動させたときのエネルギー変化を調べた。単純にするため、XYZ方向に独立に移動させた。forceが大きいものは位置を変化させたときにエネルギーが大きく変化することが予想される。
例えば(-0.03, 0.2, -0.05)のforceの場合、(0.1, 0, 0), (0, -0.1, 0), (0, 0, 0.1)の変位を与え、それぞれ(0.03, Δx), (0.2, Δy), (0.05, Δz)のエネルギー変化を得る。このときforceの大きさとエネルギー変化の関係は以下になった。

多くのサンプルで正の相関が見られた。これは当たり前のように思えるが、今回用いたGemNet-OCはforceを直接予測している(energyの微分で計算していない)ため、モデルが自発的にenergyとforceの物理的関係性を学習できていることになる。
今回の結果を特定の系に適用することで、予想と異なる結果を示したときに、モデルが誤っているのか、その系が特異的なのかを探る手がかりになる。
参考文献
[2021] Crystal graph attention networks for the prediction of stable materials
概要
正確な構造ではなく、構造のプロトタイプと組成が入力であるエネルギー予測の論文。正確な結合長をエッジのembeddingにすることで構造が未知でも入力可能とした。1500万件のペロブスカイトの仮想構造に適用し、数千の安定構造候補を抽出。

論文リンク
https://www.science.org/doi/10.1126/sciadv.abi7948
著者/所属機関
Jonathan Schmidt1, Love Pettersson2, Claudio Verdozzi2, Silvana Botti3*, Miguel A. L. Marques1
投稿日付(yyyy/MM/dd)
2021/12/3
背景
GNNの入力は構造が正確にわかっている必要があり、構造緩和後の構造が必要であったため、ハイスループットな探索が難しかった。
新規性
構造のテンプレートと組成をGNNの入力とする。
手法
モデル
- attentionを用いたGNNでモデルを構築
- エッジのembeddingはランダムに初期化される
- ノードのembeddingはMat2Vecを使用
- ノード特徴は隣接ノード、エッジ、組成をそれぞれattention weightがかかったmessageで更新
データ
- データベースはMaterials project、AFLOW、自作データベースを使用。OQMDは計算条件が他と合わないので除外。全部で200万件のデータセットを作成。
- このうちペロブスカイト構造のデータを使用。訓練データとして18万件を使用。
結果
- 訓練データのMAEは30meV/atom
- テストデータの中で2回以上登場する組成に対し、相対的な安定性を予測。エネルギー差が1meV/atomの組成でも93%の正答率。
- Materials projectのFormation energy予測ではMAEが41meV/atomであった。正確な構造なしにMEGNet等に匹敵する精度。訓練データはMPの60%を使用。
- 転移学習による精度向上も確認。おおよそ半分くらいのデータ数で同等の精度が得られる。
- バナジウムを訓練データから除外し学習したモデルにおいて、バナジウム系のデータ予測でのMAEは87meV/atomであり、外挿性もある程度高い。
- 4元系のペロブスカイトの探索では数千の候補を抽出できた。
参考文献
-
oxynitride, oxyfluorideのペロブスカイト探索
H.-C. Wang, J. Schmidt, S. Botti, M. A. L. Marques, A High-throughput study of oxynitride, oxyfluoride and nitrofluoride perovskites. J. Mater. Chem. A 9, 8501–8513 (2021). -
oxysulfideのアニオン配置
G. Pilania, A. Ghosh, S. T. Hartman, R. Mishra, C. R. Stanek, B. P. Uberuaga, Anion order in oxysulfide perovskites: Origins and implications. Npj Comput. Mater. 6, 71 (2020).
[2022] AB-INITIO POTENTIAL ENERGY SURFACES BY PAIRING GNNS WITH NEURAL WAVE FUNCTIONS
概要
機械学習による波動関数予測。従来は分子のジオメトリごとの学習が必要であったが、本モデルでは一度の学習で複数のジオメトリの波動関数が予測可能。従来モデルと同等以上の精度で高速にPESを予測できる。
論文リンク
https://openreview.net/forum?id=apv504XsysP
著者/所属機関
Nicholas Gao, Stephan Günnemann
投稿日付
2021/9/29
背景
-
機械学習によりエネルギーを直接予測するモデルは、ある点でのエネルギー値を予測するだけであり、一般的に外挿性を持たせることは困難である。そのため波動関数を直接予測することが望ましい
-
変分モンテカルロ法とニューラルネットワークを組み合わせることで多電子系の波動関数をモデリングすることに成功している。FermiNetやPauliNetなど。これらの精度を向上させるために拡散モンテカルロとFermiNetを組み合わせたモデルもある。しかし、分子の形状ごとにモデルを学習する必要があり、大量の計算が必要であった。(重み共有で計算コストを減らした系は存在するが、これも形状ごとに個別に学習が必要)
新規性
GNNのアーキテクチャと変分モンテカルロを組み合わせることで多数の形状の分子のシュレーディンガー方程式を同時に解く。これにより一回の学習でPESをモデリングすることが可能になる。
手法
-
変分モンテカルロを用いて波動関数モデル(WFModel)を繰り返しアップデートする。WFModelは電子の交換に対し符号が変化するという制約を満たすようにする。
-
WFModelを再パラメータ化するためにGNN(MetaGNN)を導入し、複数の形状に対応させる。MetaGNNは原子核の位置と電荷を入力としWFModelのパラメータを出力する。MessagePassingにより三次元のジオメトリを取り込む。
-
エネルギーを予測は回転や反転には不変であるが、波動関数は同変であるように設計する。PCAによって得られた同変座標系を使用するらしい。
-
WFModelはFermiNetをベースにし、同変座標系を導入している。
-
対称性の導入にはGNNでは相対ベクトルを用いることで取り入れることができるが、波動関数には適さない。例えばGNNでは水素原子は球対称の波動関数しか表現できないが、励起状態ではそのような対称性を有さない。そのためPCAで同変座標系を作成
-
全体のアーキテクチャ

結果
- SOTAモデルと比較して、複数の形状の分子の学習において、40倍高速かつ同等以上の精度を実現。


- ただしこれらの結果は同一のスピン配置、電子数に対する結果であり、より一般化が必要である。
参考文献
- Δ-ML:高精度計算と低精度計算の差分を学習する。
Data-efficient machine learning for molecular crystal structure prediction. Chemical Science, pp. 10.1039.D0SC05765G, 2021. ISSN 2041-6520, 2041-6539. doi: 10.1039/D0SC05765G.
[2022] Spherical Channels for Modeling Atomic Interactions
概要
球面調和関数のセットをノードに埋め込み、チャネル方向のpointwise convolutionでリッチな角度情報を取得するSperical Channel Networkを提案。同変性はm=0のみで厳密に成立するが、より多くのmを用いる(同変性は消失)ほうが高い精度を実現。OC20のSOTAを更新。
論文リンク
https://arxiv.org/abs/2206.14331
著者/所属機関
C. Lawrence Zitnick1, Abhishek Das1, Adeesh Kolluru2, Janice Lan1, Muhammed Shuaibi2, Anuroop Sriram1, Zachary Ulissi2, Brandon Wood1
1 Fundamental AI Research at Meta AI
2 Carnegie Mellon University
投稿日付
2022/6/29
背景
材料のエネルギーを予測するモデルではSO(3)同変性を付与したモデルを使うことが一般的であるが、特に小規模なデータセットにおいて制約が強く、汎化を妨げる面もある。
一方、同変でないモデルは自由度が高いものの、材料が有するべき同変性をデータ拡張により獲得する必要がある。
画像系ではCNNは並進に対し同変であるが、回転や拡大に対してはそうではない。しかし、そのようなデータにも十分対応できている。このような効果を材料系でも実現することが望まれる。
新規性
Spherical Channel Network (SCN)を提案。SCNではノードは球面調和関数のセットで表現される。
それぞれのエッジの方向に基づいてノードのembeddingを回転させ、message passingすることで角度の情報を伝達する。
同変性の制約を解くことで自由度が増し、更なる精度の向上を実現できた。
手法
ノードのembedding
球面調和関数を用いて隣接原子の角度情報をノードに埋め込む。原子間の規格化されたベクトルを入力として球面調和関数のセットに入力し、それらの線形和で特徴量を得る。
各lについて、球面調和関数は2l+1の数あるので、それより小さいlも加えて(L+1)^2の数の基底関数が存在することになる。
このembedding(球面調和関数の係数)はGNNでアップデートされる。
message passing
球面調和関数の性質から、入力を回転させることは、Wigner D-matrixで係数を変換することに対応する。

そのため隣接原子との角度情報を埋め込むには、そのベクトルをz軸方向に回転させる回転行列Rに対応する係数変換行列Dを考えれば良い。
messageを以下のように定義した。

Feは以下のような構成(左)。元素のembeddingと距離(上部)と、角度情報(下部)を掛け合わせ、全結合層で変換。最後に係数変換の逆行列を作用させ、元の次元に戻す。

同変性
同変性を保つにはFeが回転に対し不変である必要がある。これはm=0の場合にのみ成り立つ。
mが0以外のものを使うと、回転に複数の候補があり同変性は失われるが、表現力の増加により精度が向上することが確認された。(結果に記載)
なお、複数の候補の平均を取ることで疑似的に同変性を獲得することも可能であることが示されている。
モデル学習時には遠いneighborに対しては最大のLの値を小さくすることも可能。これにより精度を損なわずに計算量を減らすことが可能。
aggregate
以下のようにaggregateする。spherical gridに変換してからpointwise convolutionでチャネル方向の特徴をまとめている。これは画像で言うとフーリエ変換後の空間でチャネル方向の特徴量をまとめ、元の空間に戻すことと対応している。

energy, forcesの計算
離散点での値を積分して得る。ForcesはEnergyの微分ではなく、直接算出される。
結果
OC20 2Mで学習したところ、SCNはこれまでのSOTAをS2EFで14%, IS2REで10%更新。

単位時間あたりに処理できるサンプル数は少ないもののデータ効率が良いので相殺されるとのこと。
ablation study
- 回転を行わない(角度を取り込まない)と精度が著しく悪化。
- pointwise convolutionをなくすとせいどが悪化。
- mを0だけでなく-1,0,1を用いると、厳密な同変性が失われるが精度が向上。これよりmを多くしても精度の向上はほぼ無い。
- Lを大きくすると主にForceの精度が向上。これは角度の情報をより取り込むため。
- 回転の複数の候補に対し平均を取るとforceの精度が著しく向上する。スループットは半分になるが、学習効率が上がるので相殺される。
参考文献
[2019] High-throughput prediction of the ground-state collinear magnetic order of inorganic materials using Density Functional Theory
概要
materials projectのスピン配列のハイスループット計算。collinearの磁性パターンを特定するワークフローを開発。ベンチマーク材料の強磁性でない材料について、95%は正確に予測でき、60%は実験と同じスピン配列を予測できた。

論文リンク
https://www.nature.com/articles/s41524-019-0199-7
著者/所属機関
Horton, M. K., Montoya, J. H., Liu, M., & Persson, K. A.
投稿日付
2019/6/6
背景
-
磁性材料はスピン配列が複雑なため、ハイスループット計算の対象から除外されてきた。
-
本研究前のMaterials projectでは、ほとんどが強磁性を想定した計算しかしていなかったので、他のスピン配列も検証する。
新規性
- ハイスループットにスピン状態を決定するワークフローを提案。
手法
-
ありうる磁性パターンを数え上げ、対称性に基づいて優先づける。それらをDFT+Uで構造緩和、エネルギー計算を行う。
-
計算量の制約によりcollinearのみ検討。
-
ワークフロー全体はatomateに実装されている。
-
スピン配列の生成はpymatgenの
MagneticStructureAnalyzerに実装されている。 -
強磁性に関しては、磁気モーメントの初期値はhigh spin配置にする。これはhigh spin配置がlow spin配置にかんわされることは一般的によくあるが、low spin配置がhigh spin配置に緩和されることは無いため(エネルギーが低かったとしても)。
-
high spinに加えてzero spinの初期値も計算することで、実験との差を最小限にすることができる。
結果
-
ベンチマーク材料の強磁性でない材料について、95%は正確に予測でき、60%は実験と同じスピン配列を予測できた。
-
エネルギー値が合わないものがいくつかある原因は、擬ポテンシャルやUの値の調整をしていないため。
参考文献
[2022] Learning the crystal structure genome for property classification
概要
組成の情報を入れず、構造の特徴のみから特性に紐づく情報を抽出する試み。結晶構造の波数空間上でのXRDパターンを点群データとして扱う。入れ替えに対する不変性を満足するため、hklごとに別個に処理し、max poolingで最も影響のある点を抽出し、特性に寄与する点を抽出可能。バンドギャップには対称性が重要であるが、弾性率には電子密度が重要であることなどが判明した。
論文リンク
https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.4.023029
著者/所属機関
Yiqun Wang, Xiao-Jie Zhang, Fei Xia, Elsa A. Olivetti, Stephen D. Wilson, Ram Seshadri, and James M. Rondinelli
Phys. Rev. Research 4, 023029 – Published 11 April 2022
投稿日付
2022/4/11
背景
物性を予測する際に、組成の情報無しに結晶構造のみの影響がどうであるかを知れば、設計に役に立つ。これまでのモデルは組成情報を明示的に使用しているので、結晶構造の寄与はよくわからなかった。
新規性
組成の情報を使わず、結晶構造のみで物性を記述するモデルを作成(ただし、XRDの強度を用いているので暗黙的に組成情報は取り込まれている)。波数空間上での構造を特徴量化。
高い性能のモデルを得ることが目的でなく、構造と物性を紐付ける情報を抽出することが目標。
手法
特徴量化
- XRDパターンを波数空間で特徴量化する。これにより2Dの情報量が少ないXRDデータではなく、情報量が多い3Dのデータを用いることが可能。それぞれのhklでの強度を点群データとして扱う。
primitive cellの3D XRDを計算。波数空間で半径4π/λの球に入る範囲までにhklを制限。hklは波数空間の座標に直し、強度はlogをとった値を採用。
材料ごとにピークの数nが異なるので、nに閾値を設けた。
結晶ごとの例は以下

- 特徴量が結晶の回転やインデックスの順番に対し不変でないといけないため、データ拡張を実施。
データ
MP(139367件)について結晶系、bulk modulus, shear modulus, bandgap, energy above convex hull (E_H)を取得。
各タスクでのデータ数は以下の通り

モデル
モデルは各点ごとに4次元の特徴量を変換。この時点では各点間の相互作用は取られていないので、インデックス入れ替えに対し不変となる。変換後の特徴量をpoolingして結晶構造全体の特徴量とする。

- poolingはmax_poolingが高精度であったため採用。解釈性の面でも都合が良い。
結果
-
結晶系予測タスクでは98%の精度を達成。
-
とりうるhklの数を大きくすると、あるところまでは精度が向上し、それ以上は改善が見られなくなる。短周期すぎる特徴はあまり役に立たないことを示している。

安定性の予測は組成が大きく関わるので精度が低いとのこと。
-
入力に揺らぎを与えたデータセットφを作成し、モデルの精度を検証した。
φ2はピーク強度をランダムに0-1の値をかける(構造ごとにこの値は決めるらしい。ピークごとではなく?)。これは電子密度(元素タイプ)を変更する操作に類似。
φ3は強度を全て1にしたデータセット。対称性の情報が失われる。
これらのデータセットで学習した結果を分析することで、どの特徴が重要であるかを分析できる。
金属の分類はφ2でも精度が落ちない。元素の情報が失われているので、構造の対称性によってほとんどが決まっている。一方で弾性率はそうではなく、電子密度の情報が重要。 -
max_poolingで採用された点(予測に大きく影響を与える点)を可視化した結果が以下。

Cu, Ag, Auの同じ結晶構造でも活性化するhklは異なる。また、対称性で等価なな点を同時に抽出できており、対称性を学習していることが示唆された。
- ペロブスカイトで検証した結果、精度が悪いことが判明。これは組成の変化で物性が大きく変わるが、構造はほぼ変わらず、微妙なXRDの誤差をモデルが認識できないため。このような系には適用が難しい。
参考文献
[2022] The Open Catalyst 2022 (OC22) Dataset and Challenges for Oxide Electrocatalysis
概要
酸化物触媒の機械学習ポテンシャル構築用のOC22データセット(約1000万件)を構築。ベースラインモデルでは、異なる計算条件であるOC20との組み合わせにより(混合データ、fine-tuning)精度が向上し、よりロバスト表現が獲得できた。データセットの組み合わせ方および学習方法、スピン状態の異なるデータをどのようにモデルに取り込むかが今後の課題。
論文リンク
https://arxiv.org/abs/2206.08917
著者/所属機関
投稿日付
2022/6/17
背景
-
酸化物触媒の活性を評価するのは、様々な多型の存在、表面の複雑さ、活性サイトの特定が困難などの理由から、金属触媒より難しい。そのため酸化物触媒のデータセットは金属触媒と比較してかなり少なかった。
-
OC20データセットでは酸化物触媒は含まれていなかった。
-
OC20の提供によりモデルが著しく進化した。DimeNetでは角度項の導入、ForceNet, SpinConvではForceの直接出力(エネルギーの微分ではない)、GemNetでは三体、四体相互作用を取り入れるなど、高精度化、高速化が行われてきた。これらのモデルの活用、また更なる改良は酸化物触媒においても重要である。
新規性
-
DFTで62521系列の構造緩和(9884504件の一点計算)を実施した酸化物のデータセットOC22を作成。ベースラインモデルの精度検証も行なった。酸化物表面は、化学両論でない元素置換や空孔の導入も行い、多様性を持たせている。
-
OC20でのタスクに加え、total energyの予測タスクも追加した。より汎用的に使用できるモデル作成できることを期待している。
-
巨大なデータセット(OC20)で事前学習したモデルを用いることで、それより小規模なOC22データセットへの転移学習の有効性を検証

手法
データセット作成
-
データセットにはスラブのみの構造、スラブと吸着分子が含まれた構造がそれぞれ19086, 43435件含まれる。
-
バルク構造は4722件のAxOyおよびAxByOz構造をMaterials Projectから取得した。それぞれの組成系に対しe_above_hullが低い5構造を取得。また173件のルチル構造も取得(作成?)した。 データセットは安定性よりも化学的多様性を重視して作成されている(不安定なものを多く含む)。また、特定の組成系において著しくデータが多くなったりもしている。含有元素は以下。

-
バルクから構造をサンプリングし、ミラー指数が3以下の表面を生成。各軸長が8Å以上の表面構造をランダムに取得。さらに活性サイトの構築のためランダムに酸素空孔を生成させた(電荷中性のため反対側も同様の操作を実施)。
-
酸素生成反応中に発生する分子、ラジカルを吸着分子としてランダムに取得。特定のサイトに、分子同士が近づきすぎないように配置した(金属表面には酸素を介して配位するなどの制約はあり)。
-
計算はDFT+Uでスピンを考慮して行なった。ただし、反強磁性等は考慮しておらず、強磁性の初期スピン状態のみ考慮。全原子を緩和させるなど、OC20と計算条件が少し異なる。全計算時間は20 milion compute hoursとなった。
タスク
- S2EF-Total:構造のtotal energyとforceを予測
- IS2RE-Total:与えられた構造に対し、緩和後の構造のtotal energyを予測する。
- total energyを予測するタスクにすることで、構造緩和途中のデータも学習に使用可能になった(OC20ではスラブのみの構造と気体分子のエネルギーを全エネルギーから引いた値であったので、緩和途中はスラブのみのエネルギーの値がなく、使用できなかった)。
ベースラインモデル
- ベースラインモデルとしてSchNet, DimeNet++, ForceNet, Spin-Conv, PaiNN, GemNet-dT, GemNet-OCを用いた。PaiNNは直接Forceを予測するように改変したモデルも作成した。
学習
-
学習時のForceの係数は、構造のサイズにロバストになるように、原子数の2乗を使用した。
-
データセットと学習手順の組み合わせを3通り試した。

OC20には一応total energyも情報として持っており、その値を使用した。fine-tuningはOC20で学習したモデルに対し、OC22データのみを用いて重みの更新を行なった。今回は全ての層の重みを更新することにした。
結果
S2EF-Total

-
GemNet-OCが最も高精度あった。似たアーキテクチャであるGemNet-dTよりも高精度であったのは四体項を効果的に取り込んでいるためと予想される。
-
OODデータとIDデータの精度の差はOC20より大きかった。これはOC20はadsorption energyを用いており、一種のΔ-learningとみなせ、物理的な情報の付与により精度が向上したと考えられる。これを確かめるため、元素ごとの1原子エネルギーをデータセット全体で線形回帰し決定した。これを用いて全エネルギーから原子エネルギーの総和を引いた値を学習することで精度が向上した。このようにエネルギーの正規化スキームは今後改良の余地がある。
-
OC20とOC22両方を使って学習することでほとんどのケースで精度が向上した。DFTの計算条件が異なってもデータをミックスすることが有効であることを示している。ただしOC20データが多すぎると精度が悪化することもあった。これはOC22のデータが相対的に少なくなるため。サンプリングの戦略は今後重要になる。
-
fine-tuningでも多くのケースで精度が向上したが悪化するケースも見られた。GemNet-OCに限定し、OC22のデータサイズを変えたときのfine-tuningの効果は以下。明確に効果が出ている。

IS2RE-Total
-
直接エネルギーを予測する方法(direct)と、モデルを用いて構造緩和し、エネルギーを予測する方法(relaxation)を調査。relaxationの方が精度が高いが、200-300倍時間がかかる。
-
このタスクでもfine-tuningは有効であった。
OC22データをOC20に加える
- 逆にOC22のデータをOC20に加え、OC20の精度を検証。OC22はデータが少なく、計算条件も異なり、かつ酸化物に限ったデータセットであるにもかかわらず、精度が向上することがわかった。
展望
- 長距離相互作用をGNNモデルにどのように組み込むか。
- 計算条件が異なるデータセットをどのように組み合わせるか。
- 磁性が異なる構造の予測をどのようにモデルに組み込むか。
参考文献
[2022] Interpretable learning of voltage for electrode design of multivalent metal-ion batteries
概要
少数データの多価イオン電池について電圧予測を行い、モデルの中間層を分析することで電圧に寄与する因子を分析。酸素を含むエッジの特徴量や原子のembedding層から、共有結合半径の寄与が大きいことを特定。各元素の電圧向上に対する寄与も算出し、電極設計の指針を示した。
モデルはweb上で利用可能(http://batteries.2dmatpedia.org/)
論文リンク
https://www.nature.com/articles/s41524-022-00858-9
著者/所属機関
Xiuying Zhang, Jun Zhou, Jing Lu, and Lei Shen
投稿日付
2022/8/19
背景
多価イオン電池(MIB)への深層学習の適用は、データが少ないため進んでいない。
新規性
電極設計の指針を得るため 、DLモデルでMIBの電圧予測を行い、モデルの解釈を試みた。
手法
-
2190のLIB電極のデータで事前学習を実施。それぞれMIBのデータに対し、転移学習したモデルと転移学習なしのモデルを作成。
-
モデルはCGCNNをベースにした。
-
モデルの解釈のため、原子のembedding層と、畳み込み層を通した後のエッジ特徴量について可視化。エッジ特徴量は酸素を含むエッジ情報のみ抽出した。このエッジ特徴量を線形変換し、local voltage representationを算出。(電圧はlocal voltage representation)の平均とする。これを学習することで元素が電圧にどれほど寄与するかを調べる。

結果
- 事前学習においてLIBの電圧予測のMAEは0.32 Vであり、conventionalな機械学習モデルでの精度(0.40 V)よりも高かった。このモデルをそのままMIBに用いると、著しく精度が悪い。(下図のLi-model)

なお、conventionalな機械学習モデルを用いても、MIBに対しては精度が悪い。
- LIBのデータで学習したモデルについて、原子のembedding層をPCAした結果が以下。

伝導イオンの元素、遷移金属元素、アニオンがそれぞれ分離できている。
これは、電極電圧は構成元素種の族に強く依存することを示している。第二主成分と元素の共有結合性半径は強い線形関係を示し、電圧予測に重要な因子であることが判明した。
- 畳み込み後の、酸素を含むエッジの特徴量をPCAで可視化した結果が以下。

local voltageの分布に傾向が出ており、十分に学習できていることが分かる。また、それぞれの元素についてlocal voltageの**値を可視化した結果が以下。

遷移金属の前半の元素は一般にlocal voltageが小さく、後半は大きいことが分かった。これは元素置換の指針となりうる。
これらのlocal voltageから得られた結果は、元素のembeddingの分析から得られた結果と一致している。そのため、元素のembedddingの時点で十分な特徴を得ることができている。
- 実験値が得られている材料について検証したところ、転移学習したモデルはconventionalな機械学習モデルよりも精度が高いことが示された。

参考文献
[2022] Crystal Transformer Self-learning neural language model for Generative and Tinkering Design of Materials
概要
言語モデルを使用した無機材料の組成式生成モデルBLMMの提案。組成式の穴埋め問題を解くことで元素間の関係を捉えたモデルを構築。生成された組成式は電気的中性などの化学法則を理解できていた。既存構造の一部を置換することが可能で材料探索に適する。データセットの工夫により特性での条件付けが可能。新規材料探索では20件の候補を予測できた。
論文リンク
https://arxiv.org/abs/2204.11953
著者/所属機関
article{wei2022crystal,
title={Crystal Transformer: Self-learning neural language model for Generative and Tinkering Design of Materials},
author={Wei, Lai and Li, Qinyang and Song, Yuqi and Stefanov, Stanislav and Siriwardane, Edirisuriya and Chen, Fanglin and Hu, Jianjun},
journal={arXiv preprint arXiv:2204.11953},
year={2022}
}
投稿日付
2022/4/25
背景
-
これまでの生成モデルではモデルが何を学習したのかが不明で、獲得した知識を構造生成に活かすことができなかった。
-
Margrafらが提唱したmateriaks grammarsは専門家の知識に基づいて定義されたものであり、生成モデルのデザインスペースを絞ることができたが、化学的に要請される制限とgrammarを結びつけることは難しかった。
-
言語モデルを無機構造生成に応用した例はこれまで無い。おそらく組成式が短すぎるため。
-
本論文ではtransformerベースの言語モデルをもとに、無機材料の組成式生成や組成式変化(元素置換など)を実現するモデルBLMMを提案
新規性
- 言語モデルで無機材料の組成式を生成する
- 条件付きの生成(一部元素のみ置換など)が可能
手法
-
自己教師あり学習として穴埋め問題を解くことで原子間の複雑な関係性を得る。テキストは、展開された元素シンボルを電気陰性度順に並べ替えたものを使用する。例えばSrTiO3であれば、Sr Ti O O Oとなる。
-
組成式をスクラッチで生成するには、ブランク(#1_)から開始する。「 # 1」 はブランク、「 _ 」は末端では無いという意味。ブランクに対しE, E, E, _E_の中から1つ選び置換する。このEには元素のシンボルが入る。アンダースコアには新たなブランクができるので、この操作をブランクが無くなるまで続けることで組成式を生成する。元素置換する場合は、置換したい元素をブランクにし、モデルから置換パターンの確率を生成すれば良い。
-
BLMMは以下のように学習する

ブランクありの組成式を用意し、transformerのエンコーダで処理する。ブランク部分を変換し、どのブランクが最初に処理されるかを決定する(blank selection network)。次にelement selection networkでどの元素で置換するかを決定する。2つのネットワークで得られているembeddingを結合したのちMLPで変換し、どの置換タイプが適切か決定する。
組成式に対しブランク数tと置換順序σをサンプリングする。σに従い、それぞれのブランクに対しアクションaを予測する。ロスは以下で表される。

- データセットはICSD, MaterialsProject, OQMDから取得。電気的中性を満たさないものや、電気陰性度に偏りがあるものを含むデータセットと、満たすもののみのデータセットを作成。
結果
-
生成した仮想組成式の分布は既存手法(MATGAN)よりも、実際に近い分布を形成した。

-
OQMDデータセットでは、生成組成式のうち、電気的中性が保たれたもの(CN)は89.7であり、化学的なルールを学習できていた。また、CNが保たれていないものを含むOQMDデータセットで学習した場合、生成組成式のうちCNを満たすものは69.9%であったことから、データセットの質が重要であることがわかる(CNを満たす組成式のみのデータセットの方が40%少ないのにも関わらず。)。
-
BLMMは学習データに近い仮想的な組成式を生成しやすい(元素置換に適している)のに対し、MATGANは新規組成を探索するのに適している傾向があった。
-
いずれのデータセットでもユニークな組成の生成割合は、100万組成を生成した時点でも50%を超えた。
-
学習データの再現率は2元系で98.3%, 3元系で80.0%, 4元系で45.3%であり、MATGANと比較して高い値を示した(5-6倍)。これはBLMMが学習データに近い分布を生成することと一致している。ただしテストデータの再現率は4元系では29.1%に下がる。
-
異なるエポックごとで保存したモデルを用いて、CNやBalanced Electronegativity(BE)が満たされる割合を調査したところ、エポックを増やすごとにほぼ単調にCNやBEを満たす組成が増加したため、学習が進むことで化学の法則を理解できるようになっていることが分かった。生成された組成式を見ると、1エポックではほぼランダムに生成されていたものが、カチオン・アニオンをバランスよく含みCNを満たすように生成されていく様子が見られた。50エポック程度では同じ元素が隣にならないこともあったが、さらに学習すると改善した。これにより電気陰性度も理解していることが分かる。

- 様々な既存組成の一部をマスクし、置換組成を予測した結果が以下。マスクした部分は高い確率で予測され、かつ新規構造の予測もできた。(Sr3CrN3は報告されているがデータベースには含まれない)

-
バンドギャップが1.98 eV以上の組成29772件で学習し、生成された組成のバンドギャップ(予測モデルを使っているので値はアバウト)
を予測。生成された組成のバンドギャップは、全データで学習した場合と比較して大きいものが多く、バンドギャップが大きい組成というものを暗黙的に理解していることが分かった。このようにデータセットを工夫することで、物性で条件付けすることも可能。 -
3, 4元系の組成10万件を生成し、組成からの形成エネルギー予測モデルで安定な組成を抽出し、安定なものをTCSP(テンプレートベースの構造予測ツール)で構造を予測したところ、20件の新規材料を発見した。
参考文献
-
MATGAN:
Yabo Dan, Yong Zhao, Xiang Li, Shaobo Li, Ming Hu, and Jianjun Hu. Generative adversarial networks (gan) based efficient sampling of chemical composition space for inverse design of inorganic materials. npj Computational Materials, 6(1):1–7, 2020. -
TCSP(テンプレートベースの構造予測ツール):
Lai Wei, Nihang Fu, Edirisuriya Siriwardane, Wenhui Yang, Sadman Sadeed Omee, Rongzhi Dong, Rui Xin, and Jianjun Hu. Tcsp: a template based crystal structure prediction algorithm and web server for materials discovery. Inorganic Chemistry, 2021.
[2021] Physics guided deep learning generative models for crystal materials discovery
概要
物理に基づいたデータ拡張、ロス関数、ポストプロセスを用いたGANにより現実的な構造を多く生成できるようになった。
シンプルな系であるが、実際に安定な構造を生成することに成功。

論文リンク
https://arxiv.org/abs/2112.03528
著者/所属機関
Yong Zhao, Edirisuriya MD Siriwardane, Jianjun Hu1* 1Department of Computer Science and Engineering University of South Carolina
投稿日付(yyyy/MM/dd)
2021/12/7
詳細
-
データ拡張は
base atomのサンプル方法を変える。これはこのモデル特有の操作っぽく、あまり汎用的ではない気がする。 -
ロス関数に、原子間を遠ざけるように働く関数を加える

これだけだと際限なく大きくしてしまうので、格子パラメータのコサイン類似度もロスに加えることで、原子間距離が大きくなりすぎないようにする。
-
ポストプロセスは、対称操作により近い位置に来てしまった原子群を階層的クラスタリングでまとめる
-
以下のような構造が生成できた。

参考文献
- 元となったモデル(CubicGAN)
Zhao, Y.; Al-Fahdi, M.; Hu, M.; Siriwardane, E.; Song, Y.; Nasiri, A.; and Hu, J. 2021. High-throughput discovery of novel cubic crystal materials using deep generative neural networks. Advanced Science.
[2022] A deep learned nanowire segmentation model using synthetic data augmentation
概要
合成画像のデータセットのみを用いて実データのセグメンテーションを行なった論文。ロッド形状のオブジェクトに対し、optical densityを付与し、実データの信号強度を模倣。STXMやSEMデータで高い性能を示した。
論文リンク
論文:
https://www.nature.com/articles/s41524-022-00767-x
Webアプリ:
https://share.streamlit.io/linbinbin92/V2O5_app/V2O5_app.py
データ、コード:
https://github.com/linbinbin92/V2O5_app/tree/master
著者/所属機関
@Article{lin2022deep,
title={A deep learned nanowire segmentation model using synthetic data augmentation},
author={Lin, Binbin and Emami, Nima and Santos, David A and Luo, Yuting and Banerjee, Sarbajit and Xu, Bai-Xiang},
journal={npj Computational Materials},
volume={8},
number={1},
pages={1--12},
year={2022},
publisher={Nature Publishing Group}
}
投稿日付
2022/4/28
背景
- 粒子のセグメンテーションは教師データを大量に集めることが困難
新規性
合成データのみを使ってモデルを学習
手法
合成データセット
- 三次元の箱にロッド形状のオブジェクトを作成。これにoptical densityの値をボクセルに付与する。縦方向に合計、正規化することでbinary画像を得る。エッジはガウシアンフィルターで正規化。

モデル
通常のMask R-CNNモデルを使用。backboneはResNet-50のFPN。COCOデータセットで事前学習した重みを初期値にした。

結果
- 複数のタイプの実データで高い性能を示した。
合成データの予測精度

ほぼ完璧な予測。ちなみにwatershedg法ではロッドの分離はうまくいかず本手法の優位性が示された。
X-ray pytchographyデータ

青がTP, 赤がFN, 緑がFP。概ね正しく検出できている。
STXMデータ

こちらも予測精度は高い。FPがあるが、人間のアノテーションが間違っているせい。
SEMデータ
- SEMデータにはoptical densityの情報がないので、今回の中で一番難しいタスク。粒子の検出はうまくいっているが、大きな領域のFPが出てしまっている。これはより多くのパターンの合成データを学習させることで改善が可能と主張している。

- 本手法は他の形状の粒子にも容易に適用可能。
- 今後は光のぶれや角度の影響を模倣するため、データにノイズを加えて学習することで精度が向上するか検証予定。
参考文献
[2022] Super-resolving microscopy images of Li-ion electrodes for fine-feature quantification using generative adversarial networks
概要
SRGANでSEM画像を超解像し、LiBの正極活物質のクラックに関してのセグメンテーションを実施。低解像度のSEM画像を単純にアップサンプリングした場合と比較して、SRGANで生成した高解像画像を用いることで精度が向上。perceptual lossはSEMの超解像にも有効であり、鮮明な高解像画像の出力が可能であった。低解像と高解像のペアが異なる画像の場合はCinCGANのアーキテクチャを用いると良い。
論文リンク
https://www.nature.com/articles/s41524-022-00749-z
著者/所属機関
Orkun Furat, Donal P. Finegan, Zhenzhen Yang, Tom Kirstein, Kandler Smith and Volker Schmidt
投稿日付
2022/4/13
背景
- 活物質のSEMは不均一なことが多く、特徴を定量化するためには複数粒子を捉える大きな領域での分析が必要であるが、SEMの解像度が悪くなり、細かな特徴の分析(クラックの分析など)が正しく行えないことがある。
新規性
-
GANを用いたモデルで正極活物質のSEM像の超解像を実行(SRGAN)。NMC粒子のクラックの分析を実施。
-
これまでに報告されたSEM像の超解像モデルとのベンチマークを実施
手法
GANのアーキテクチャ
- SRGANをベースにモデルを作成。
- 生成器は16ブロックのSRResNetを使用。PReLUをReLUに変更し、バッチ正規化を除去(今回のデータでは無い方がよかったらしい)。低解像度のSEMから縦横a倍(今回は2.5倍)の高解像度データを生成する。

最適化
-
perceptual lossを導入。これは学習済みの画像識別器に、画像のペアを入力し、途中の特徴量のMSEを取る。画像同士のMSEを取ると、ぼやけた画像を出力する(ピクセルずれでもlossが大きくなるので、スムーズな画像を出力しようとする)ため、代わりにperceptual lossを使用する。
-
学習には低解像度の画像と高解像度の画像のペアが必要であるが、ペアがない場合は高解像度の画像をダウンサンプリングして低解像度の画像を作ることも可能。ただし、実験で得られる低解像度のデータと性質が同じでないことから性能は低下する。そのような場合はCycleGANでの超解像を検討すると良いとのこと。
-
データはtrain, val, testでそれぞれ24, 5, 4ペアの画像を使用。(ここから画像を切り出して学習)。画像のペアはOpenCVの
matchTemplate()関数を使用。 -
様々なモデルで学習しベンチマークを行なった。

結果
- それぞれのモデルでの超解像データは以下

多くのモデルはノイズ除去ができているが、SRResNet2はノイズが乗っている。SRResNet2は高解像度画像とそれをダウンサンプリングして得られる低解像度画像のペアで学習しており、実験で発現するノイズの性質を捉えられなかったためであると考えられる。
低解像度画像と高解像度画像の異なるペア(1つのSEM画像の低解像度画像と、もう1つのSEM画像の高解像度画像)で学習させたCinCGANではSRResNet2を上回るパフォーマンスが得られた。
perceptual lossを加えたSRGANは最も高精度であり、SEMの超解像には有効であると考えられる。
- それぞれのモデルが出力した高解像画像を用いてクラックのセグメンテーションを行った結果は以下。


実験の低解像度から得た結果(c)よりも、超解像で得られたデータの方が、実験の高解像度(b)から得られた結果(b)に近い。
SRGANは低解像度のデータをアップサンプリングするよりも、全ての指標で良い結果を示した。
参考文献
[2021] SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects
概要
機械学習ポテンシャルにおいてinductive biasをふんだんに盛り込んだSpookyNetの提案。チャージアップやスピン状態などを明示的にモデルに組み込むことで精度向上。また近距離の反発力やファンデルワールス力などのポテンシャルの差分を学習することでPESが高精度になっている。
論文リンク
https://www.nature.com/articles/s41467-021-27504-0
著者/所属機関
Unke, Oliver T and Chmiela, Stefan and Gastegger, Michael and Sch{"u}tt, Kristof T and Sauceda, Huziel E and M{"u}ller, Klaus-Robert
投稿日付
2021/12/14
背景
- これまでのモデルは電子の状態(total chargeやスピン状態)を考慮しておらず、これらの影響が大きい系で正確に予測できなかった。
新規性
- 電子の自由度や非局在性を明示的に取り入れたアーキテクチャを提案。
手法
-
原子番号、座標、電子数、スピン状態を入力とする。座標以外の特徴量を変換し結合したものがノード特徴量となる
-
角度情報は基底関数(Bernstein polynomialsと球面調和関数)により取り入れられる。(角度を明示的に計算しない)
-
invariantなエネルギーを予測するだけなので、equivariantな特徴量は計算コストの観点からinvariantな量に変換して計算する。
-
原子核の反発(Ziegler-Biersack-Littmark stopping potential)、長距離の電子相互作用、ファンデルワールス力(D4分散力補正)とDFTでのエネルギーとの差分を学習する。
-
localとnonlocalに分けて寄与分が計算される。

localはs,p,d軌道を再現するためのものでnonlocalは非局在化した電子をモデル化するためのもの。 -
全体のアーキテクチャは以下

結果
- total chargeやスピン状態を取り入れることで、DFTを高精度に再現

- nonlocalの寄与を入れることで2体間ポテンシャルの精度が向上

- ベンチマーク結果

参考文献
-
電子密度の情報を入れる
Ko, T. W., Finkler, J. A., Goedecker, S. & Behler, J. A fourth-generation high- dimensional neural network potential with accurate electrostatics including non-local charge transfer. Nat. Commun. 12, 398 (2021). -
スピン状態を入れる。
Zubatyuk, R., Smith, J. S., Leszczynski, J. & Isayev, O. Accurate and transferable multitask prediction of chemical properties with an atoms-in- molecules neural network. Sci. Adv. 5, eaav6490 (2019).
[2021] Element selection for crystalline inorganic solid discovery guided by unsupervised machine learning of experimentally explored chemistry
概要
無機材料の合成可能性予測。入出力を元素ペアの特徴量としたVAEを学習。元素ペアに対するVAEの再構築誤差により、合成可能性を評価。アニオン2種の4元系Li固体電解質の探索を実施し、Li3.3SnS3.3Cl0.7の新規相合成に成功。

論文リンク
https://www.nature.com/articles/s41467-021-25343-7
著者/所属機関
Andrij Vasylenko, Jacinthe Gamon, Benjamin B. Duff, Vladimir V. Gusev, Luke M. Daniels, Marco Zanella, J. Felix Shin, Paul M. Sharp, Alexandra Morscher, Ruiyong Chen, Alex R. Neale, Laurence J. Hardwick, John B. Claridge, Frédéric Blanc, Michael W. Gaultois, Matthew S. Dyer & Matthew J. Rosseinsky
投稿日付
2021/9/21
背景
無機材料の合成可能性予測は多数の要因が関連する複雑な問題であり、実現が困難であった。
新規性
元素ペアの特徴量を入出力としたVAEを作成。個々の組成や構造に着目するわけではない。VAEは合成可能なものだけに焦点を当てている。これは材料分野では望ましい。合成不可な材料は単に合成法が悪いだけの可能性を含んでおり、完全に正しいラベルとは言えないため。
手法
- 元素特徴量37個を抽出。元素ペアの特徴量を全て結合し入力とする。
- VAEは通常のものを使用
- 元素並び替えのデータ拡張を行う
- VAEの再構築誤差により、元素ペアの合成可能性をランクづけする
- 提案された元素ペアに対し、GA等で構造を生成し安定なものを抽出
- 真空中の固相法で合成
結果
-
ICSDの登録材料をVAEで評価すると再構築誤差が0.5以下が8割で、VAEはうまく学習できている
-
再構築誤差が小さかったLi-Sn-S-Cl系について、エネルギーが低かった相図中の6点を合成した結果、2点で新規相が得られた。
-
計算によるスクリーニングでは出てきにくい中途半端な組成(Li3.3SnS3.3Cl0.7)を単相で合成。
-
ウルツ鉱型でアニオンはランダムに配置。Liの一部は八面体サイトに位置する。

-
3.2E-05 S/cmでLi金属に対し(Li4SnS4のカチオン置換体と比較して)安定。
-
Liイオン伝導パスはこれまでのhcp格子では見られないパターンであり、単一のアニオン系のパターンとは異なる。
参考文献
合成可能性予測
-
Haynes, A. S., Stoumpos, C. C., Chen, H., Chica, D. & Kanatzidis, M. G. Panoramic synthesis as an effective materials discovery tool: the system Cs/Sn/ P/Se as a test case. J. Am. Chem. Soc. 139, 10814–10821 (2017).
-
Canfield, P. C. New materials physics. Rep. Prog. Phys. 83, 016501 (2019).
-
Xia, Z. & Poeppelmeier, K. R. Chemistry-inspired adaptable framework
structures. Acc. Chem. Res. 50, 1222–1230 (2017). -
Wong-Ng, W., Roth, R. S., Vanderah, T. A. & McMurdie, H. F. Phase
equilibria and crystallography of ceramic oxides. J. Res. Natl Inst. Stand. Technol. 106, 1097–1134 (2001). -
Ward, L., Agrawal, A., Choudhary, A. & Wolverton, C. A general-purpose machine learning framework for predicting properties of inorganic materials. Npj Comput. Mater. 2, 16028 (2016).
-
Schmidt, J. et al. Predicting the thermodynamic stability of solids combining density functional theory and machine learning. Chem. Mater. 29, 5090–5103 (2017).
-
Liu, Y., Zhao, T., Ju, W. & Shi, S. Materials discovery and design using machine learning. J. Materiomics 3, 159–177 (2017).
-
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. & Walsh, A. Machine learning for molecular and materials science. Nature. 559, 547–555 (2018).
[2022] Open Challenges in Developing Generalizable Large Scale Machine Learning Models for Catalyst Discovery
概要
汎用的な機械学習ポテンシャル構築に対する課題と展望。モデリング方法(1. forceを直接予測するか、エネルギーを微分して得るか, 2. 緩和構造を直接予測するか、最適化アルゴで得るか)やデータセット作成法、構造緩和タスクの評価指標などが確立されておらず、まだまだ課題が多い状況。
論文リンク
https://arxiv.org/abs/2206.02005
著者/所属機関
@Article{kolluru2022open,
title={Open Challenges in Developing Generalizable Large Scale Machine Learning Models for Catalyst Discovery},
author={Kolluru, Adeesh and Shuaibi, Muhammed and Palizhati, Aini and Shoghi, Nima and Das, Abhishek and Wood, Brandon and Zitnick, C Lawrence and Kitchin, John R and Ulissi, Zachary W},
journal={arXiv preprint arXiv:2206.02005},
year={2022}
}
投稿日付
2022/6/4
背景
- 機械学習ポテンシャル(NNP)の大規模データセット(OC20)により様々な元素に適用可能なユニバーサルなモデルへの道が開かれた。しかし課題がいくつかあり、本論文で紹介する。

新規性
手法
結果
モデリングの動向
-
初期はハンドクラフトな特徴量を用いていた。これは距離、角度の特徴量で、構造変化に対しinvariantな設計になっている。
-
ここ数年でGNNにより自動で特徴量を学習するようにシフトした。原子をノード、距離をエッジとする。このアプローチは従来大量のデータを必要とするが、NequIPは100サンプル程度でも素晴らしい性能を示した。
-
OC20が公開されてから、新しいモデリングアプローチが急激に増えた。現在のSOTAはGemNet-OCモデル。他にもグラフを並列化することで複数のGPUで10億パラメータのモデルの学習が可能になっている。

- OC20データで事前学習したモデルを用いて、ドメイン外のデータセットに対し転移学習することで高い精度を得た例もある。
モデルの性能
-
OC20データセットで学習したモデルは、元素と吸着分子のタイプにより精度が均一でないことを確認している。NonmetalsとMetaloidsは同程度のデータ数であるが、Nonmetalsの性能はMetaloidsに大きく劣っている。
-
吸着分子の原子数が大きくなると自由度が上がり予測が難しくなると考えられるが、モデルの性能は吸着分子の原子数によらなかった。吸着分子にNかOが存在するとエラーが大きくなる傾向があった。
モデリングのトレードオフ
-
物理的にはforceはenergyを原子位置で微分したものである必要がある。このモデリング方法ではforceは回転に対しequivariantであることが保証され、物理的に正しいforceが得られる。一方でモデルが消費するメモリ量、計算量は一般的に2-4倍程度になってしまう。また、学習が不安定になる場合があった。
-
直接forceを予測するようなモデリング方法では、構造緩和のタスクにおいて、GenNet-dT, Spinconvモデルでは、forceをenergyの微分値として得た場合よりも精度が向上した。また、それらのモデルを用いて構造緩和した後のenergy, forceも、微分値で得たモデルで緩和した場合よりも精度が高かった。エネルギー保存則を厳密に満たす必要がないタスクでは直接forceを予測する方が良い。
緩和後のエネルギー、構造予測
- 緩和後の構造予測では、energy, forceをモデルで算出し最適化アルゴリズムとともに構造緩和する方法(緩和法)と、緩和前構造から緩和構造を直接予測する方法(直接法)がある。緩和法では200-300回の評価が必要であるのに対し、直接法では1回の評価で済む。さらにデータ数が緩和法の1/300程度であるにも関わらず、直接法はドメイン内データにおいて緩和法に匹敵する精度が得られた。一方でドメイン外データに関しては緩和法が優れていた。
局所安定の検出のためのmetrics
-
緩和後構造予測タスクではmetricsが明確でない。予測構造は、正解と同じ局所解に到達、異なる局所解に到達、局所解に到達しないの3種類がある。これを区別するため、正解構造との距離ベースの指標を用いている(ADwT)。このとき、任意の座標系に対応するために、絶対値でなく、初期値からの変化量で定義している。
しかし、この指標では対称操作により同一になるものや、他のリーズナブルな局所解のものを悪く評価してしまう。そのため、緩和後の構造の力を評価し、0に近いものがどの程度あるか(AFbT)を追加することでより好適なmetricとなる。より正確なmetricとしては、MLで得られた緩和後構造をDFTで緩和し、収束までのステップ数を指標とするものがある。しかしこれらはDFT計算のため、非常に計算コストが高いmetricsである。 -
直接法では緩和法と比較して、AFbTが著しく悪いことが判明した。そのため直接法は構造としては正解と似たものを出力するが、反発力を無視したような配置になっていることが判明した。これは少しの位置の変化でforceは大きく異なることに由来する。
-
計算コストの低いADwTではthresholdを小さくしても、より正確な指標だが高計算コストのAFbTを代替する指標にはならないことが判明した。そのため、計算コストが低く、正確に緩和タスクの性能を評価する指標が今後必要。
追加データ
- OC20では追加データとしてMDデータ(MD)、摂動データ(Rattled)が与えられている。force予測では追加データによりDimeNet++では悪化、GemNet-OCでは良化した。一方で緩和構造予測ではAFbTはいずれも明確に向上した。

いくつかの要因(大きな変位の構造はは予測に重要でないなど)考えられるが、どのようにデータセットを作るべきかは明確でない。active learningが有望な方向性である。
今後取り組むべき課題
-
データセットの分割方法
データをどのような括りで分けると良いか明確ではない。少ないデータセットで良いモデルを得るために重要。 -
不確実性の定量化、能動学習
従来の不確実性評価手法をOC20の巨大データセットに適用したときの効果を調べることが重要。
また、データ収集、データ拡張に能動学習をどのように適用すべきか検証が必要。 -
モデルの計算効率
equivariantな性質の利用、蒸留などでモデルの計算コストを減らすことが重要 -
データ増強
OC20はデータが多すぎて、少しの量のデータを増やしても精度はあまり変化せず影響が分からない。ANI-1などの大規模データを追加し検証することは有効だが、異なるDFT計算条件でのデータをどのように結合するかが最大の課題になっている。 -
force出力方法
OC20では直接forceを予測することが良かったが、MD17ではそうではなかった。このようにどちらの手法で出力すべきか明確でない。 -
物理のドメイン知識の導入
特にデータが少ない領域において物理のドメイン知識導入は有効である。Bader chargeなどのデータを有効に用いる方法が必要である。
参考文献
-
GemNet-OC:
Gasteiger, J.; Shuaibi, M.; Sriram, A.; Gu ̈nnemann, S.; Ulissi, Z.; Zitnick, C. L.; Das, A. How Do Graph Networks Gener- alize to Large and Diverse Molecular Sys- tems? 2022; https://arxiv.org/abs/ 2204.02782. -
OC20モデルを転移学習
Kolluru, A.; Shoghi, N.; Shuaibi, M.; Goyal, S.; Das, A.; Zitnick, L.; Ulissi, Z. W. Transfer Learning using Attentions across Atomic Systems with Graph Neural Networks (TAAG). The Journal of Chemical Physics 2022,
[2022] Powder X-Ray Diffraction Pattern Is All You Need for Machine-Learning-Based Symmetry Identification and Property Prediction
概要
XRDの入力のみを用いたモデルで、特定のタスクにおいてGNNベースの手法よりも高精度を実現。対称性を高度に取り込むことから、結晶系予測や、対称性が絡んだ物性の予測に好適。電子分布を取り扱う類似性から、DFTで計算される物性の予測に強いと主張。
論文リンク
https://onlinelibrary.wiley.com/doi/full/10.1002/aisy.202200042
著者/所属機関
投稿日付
2022/5/22
背景
XRDを入力として用いたモデルでは対称性の予測や相同定などはあるが、物性の予測は研究例が無い。
また、一部の組成系に限られており、汎用的に使えるかどうか分かっていない。
新規性
XRDをそのまま入力としたモデルを作成。(CNNベース、transformerベース)ICSDとMPの全データでの汎化性能を調査
手法
データセット
-
ICSDとMPの全構造を取得。体積が10,000Å^3を超えるものは除外。この制約においては5°以下のピークは出てこないと考えられるので、5-86.91°までの8192個の1次元値を入力にした。データは全部で18946(ICSD), 139207(MP)件。
-
MPのXRDデータは、実験データに近くなるような操作を加えた(ランダムなバックグラウンドなど)。
モデル
- 1次元CNN(FCN), transformer(T-encoder)、VAEのモデルを作成。CNNは組成の情報も加えたモデル(FCN-MLP)も同時に作成。
FCNはCNNの出力をそのまま用いている(MLPに流さない)。著者の以前の研究ではCNNの出力をMLPに流して出力を得ていたが、MLP無しでも精度が同等であることを確認しており、パラメータ数を減らすことができるとのこと。

-
FCNは13のCNN層、maxpooling, dropoutで構成。
-
T-encoderは2個のmultihead attentionブロックを使用。XRDデータは64のパッチに分割して入力とした。ViTとは異なり、それぞれのパッチに変換は行わず直接入力とした。
-
FCN-MLPでは、XRD処理部(CNN)と、組成処理部(MLP)で構成。組成処理部の入力は、組成比を正規化し、multi-hotにしている。それぞれの処理部での出力を結合し、別のMLPを通して出力を得る。
-
対称性予測で学習したFCNを初期パラメータとし、VAEを学習。XRDのデータ分布はガウス分布ではないことから、サンプリングには指数分布とガウス分布の両方を採用。
結果
- ICSDデータの結晶系の同定ではFCNで92.1%の精度を実現。一方でT-encoderの精度は低く、79.6%であった。

MPデータでは少し下がり、FCNで80%程度。一般的なXRD解析ツールを用いても80%程度が限界であることから、高い精度を得ていると言える。
専門家の精度はこれより上回るものの、本手法は高速に処理できることがメリットであると主張している。
ICSDで予測精度が高いのは以下の理由。
- 重複したデータが存在している
- MPは完全占有であるが、実際は部分占有である可能性があり、対称性が変わる。(MPの対称性はICSDよりも低いところに分布している。)
-
T-encoderの精度が低いのは、パラメータ数を増やすことができなかったため。データ数が増えるとFCNを超える可能性はある。あるいは何らかの自己教師あり学習を行う。
-
ベースラインとして使用したCGCNNではFCN, T-encoderのどちらよりも精度が低かった。これはCGCNNでは対称性をうまく取り込めていないことを示す。おそらく局所的な情報はよく反映されるが、長距離の周期構造の情報は完全には伝わっていないため。
-
FCNでは物性予測の精度は低かったが、組成情報を入れ、FCN-MLPにすることで精度が劇的に改善。CGCNNの精度と同程度。

バンドギャップなど、対称性にある程度関わる物性はCGCNNよりも良い性能を示した。
-
組成のみのモデルで同様に物性の予測をすると、バンドギャップの精度は悪化したが、形成エネルギーは悪化しなかった。これはエネルギーは組成で多くの情報を取り込めるが、対称性は取り込めず、XRDが必要であることを示す。
-
XRDは三次元の電子密度の情報を1次元に射影しているので、構造の完全な情報を取り込むことはできない。しかし構造がなくてもXRDパターンがあれば本モデルを使用可能。また、電子の空間情報を取り込んでいるので、DFTで計算された物性を予測する際には有効に働くであろうと主張している。
-
結晶系ごとに精度を見ると、triclinicなど対称性が悪いものは精度が悪かった。
-
VAEのembeddingではうまく結晶系を分けることが可能であった。学習時のロスの設計により分布の形状が異なる。

- VAEでXRDパターンを生成すると、妥当なパターンが得られることが分かった。しかし、新規構造の発見につながるような品質ではない。また、U-Netの構造を取り入れることで品質の向上が見られた。
参考文献
[2022] Featurizations Matter: A Multiview Contrastive Learning Approach to Molecular Pretraining
概要
SMILES, fingerprint, 2D/3Dグラフの4つの表現をattention層で適応的に組み合わせる事前学習手法MEMOを提案。事前学習はそれぞれの特徴量をアンカーとした対照学習を実施。8個中7個のデータセットでSOTA更新。得られたattentionの重みを用いて下流タスクごとの特徴量寄与度を分析可能。
論文リンク
https://openreview.net/forum?id=Pm1Q1X3avx1
著者/所属機関
Yanqiao Zhu and Dingshuo Chen and Yuanqi Du and Yingze Wang and Qiang Liu and Shu Wu
投稿日付
2022/5/21
背景
- 分子の表現学習において、SMILESや2D/3D表現でのベクトル化が用いられてきたが、そららを組み合わせた効果は分かっていなかった。fine tuning時も単一の特徴量が使われており、組み合わせの議論がなかった。
新規性
-
4つの分子表現(SMILES、2D/3D構造、fingerprint)を用いて事前学習を実施するフレームワークMEMOを提案。attentionを用いており、それぞれの表現の寄与度の分析が可能。対照学習を用いた自己教師あり学習で事前学習を行う。
-
他の事前学習手法との差は以下。

手法
分子表現
-
GINモデルで2次元の分子構造を表現する。
-
SchNetで3次元の分子構造を表現する。
-
ECFP4のスキームで分子のfingerprintを得る。スパースなベクトルのためembedding層、self-attention層を通して表現をアップデートする。これをsum pooling後に線形変換し、fingerprintのembeddingを得る。
-
学習済みのRoBERTaを用いてSMILESを変換する。
-
上記の4つの特徴量の加重平均をとり、全体の特徴量とする。この時の重みはattention層から算出する

対照学習
得られた特徴量を用いて対照学習で事前学習を行う。InfoNCEの目的関数を使用。アンカーは上記4つの特徴量とし、全体の特徴量との類似度を正例と負例に対し計算する。

結果
- MEMOは他のSSL手法と比較して、ほぼ全ての下流タスクにおいて最も高い性能を示した。

また、事前学習無しのモデルと比較しても精度が向上しており、事前学習において、下流タスクを解くための知識を獲得できていると言える。
- 各下流タスクにおいて、attentionの重みを可視化した結果が以下。

分子の特性への寄与は3次元の細かな配置よりも、2次元の分子の配列の方が大きく、化学的な知見とも一致している。2次元は官能基などの基本的な情報が含まれており、これが大きく物性を左右する。
また、SMILESは分子の配列を記述してはいるが、表現力が不十分であり、2次元のグラフ特徴量の方が有用な特徴が得られる可能性が高いことが示唆されている。
fingerprintは局所的な特徴しか得られないので、重みは低い傾向にあった。
- attention層を取り除いたモデル(MEMO-Max)、attentionでなく平均を取ったモデル(MEMO-Mean)、finetuningを無くしたモデル(MEMO-Freeze)を作成し、ablation studyを行なった結果が以下。

attention層無しでは精度が低く、特徴量を動的に選択することが重要であることを示している。finetuning無しでも、attention無しのモデルより高い精度が得られているため、attentionで分子の良い表現が得られていると言える。
参考文献
[2022] Evaluating Self-Supervised Learned Molecular Graph Representations
概要
分子の事前学習において、embeddingがどのような特徴を捉えているか調査。事前学習により大域的な情報及び構造のサブユニットの認識性能が向上し、下流タスクの精度が向上することが示唆。over-smoothingやdimension collapseは起こっていないことを確認。一方で局所的な情報はむしろ失われていることが判明。
論文リンク
https://openreview.net/forum?id=LeJC_Mf5rx-
著者/所属機関
author={Hanchen Wang and Shengchao Liu and Jean Kaddour and Qi Liu and Jian Tang and Matt Kusner and Joan Lasenby},
booktitle={ICML 2022 2nd AI for Science Workshop},
投稿日付
2022/6/15
背景
現実的に収集可能なデータ数は限られているので自己教師あり学習(SSL)が注目されているが、モデルが何を学習し、下流タスクにどう関係しているかよく分かっていない。
新規性
分子の事前学習に関して、embeddingがどのような特徴を捉えているかを調査
手法
-
GNNのSSLは過去に報告された手法で実施。バックボーンとしてGraph Isomorphism Network (GIN)を使用。
-
10種類のSSLを実施。EdgePred, InfoGraph, GPT-GNN, AttrMask, ContextPred, G-{Contextual, Motif}, GraphCL, JOAO{・, v2}を用いた。実験条件は元の論文に従った。
-
SSLのデータセットはGEOMからランダムに5万件抽出し作成した。
-
SSL後のモデルから、embeddingを抽出し、それを元にプローブタスクを実施。プローブモデルはSSLで得られた表現が、有益な構造情報をエンコードできているか調べるためのモデル。embedding部分は固定し、浅いネットワークをそれぞれの評価指標で学習させる。今回は1層のMLPを用いた。
-
SSL後のembeddingについて、ノードレベル、ペアレベル、グラフレベルでの分析を実施。
結果
- 事前学習での精度向上は以下の通り

いずれのSSLでも精度が平均的に向上している。
また、それぞれ事前学習したembeddingを用いて、ノードレベル、ペアレベル、グラフレベルの評価指標の予測精度を調べた結果が以下。

事前学習したモデルはグローバルな(グラフレベルの)トポロジーに関連する情報の予測精度が向上した。一方で、局所的な情報は、事前学習なしの方が予測精度が高かった。また、全ての指標に有効な事前学習方法はそんざいしないことが分かった。
G-Contextualなどでは、事前学習での目的関数と、予測対象の量との結びつきが強く、高い精度が得られた。そのため、予測する物性と関連する(かつ簡単に算出可能な)量を事前学習のターゲットとするのが良いと思われる。
contrastiveベースの事前学習(JOAOなど)はグラフレベルの予測が期待されるほど向上しなかった。
- 事前学習したモデルにおいて、それぞれのサブユニットの個数を予測するタスクの精度は以下の通り

事前学習ありのモデルは分子のサブユニットの個数を予測する精度が高かった。また、サブユニット予測タスクの精度と、下流タスクの精度の相関は高かった。さらに、サブユニットの個数を特徴量として、決定木ベースの機械学習モデルで物性予測を行うと、精度が高かった。このため、事前学習による精度向上の一因は、サブユニットをうまく認識できることに由来していることが示唆された。
- 画像でのSSLでよく観測されてしまう、dimension collapseについて調査。embeddingの特異値を計算し、プロットした結果が以下。

高次元でも情報を保持しており、dimension collapseは起こっていないことが確認された。
- 分子の正例ペア(物性のラベルが全て同一)と負例ペアをランダムに1万個取得し、embedding上でのコサイン距離を算出した結果が以下。

事前学習により、正例ペアと負例ペアを区別することが可能になっていることが分かる。over-smoothingが起こると、これらのペア同士は区別できないことから、事前学習によりover-smoothingを緩和できることがわかった。
- 事前学習で得られたembeddingについて、ノードの次数で色分けしたt-SNEの結果は以下。

事前学習により局所的な情報が失われていることが分かる。
embeddingをとる場所を変えたときの精度変化は以下の通り

前半のembeddingほど、局所的な情報(node)を持っており、後半に行くにつれて大域的(graph)な情報を保持していることが分かる。(GNNの機構的にそうあるべき)
- readoutも目的に応じて適切に選択する必要あり。
参考文献
[2021] Predicting synthesizability of crystalline materials via deep learning
概要
結晶構造の合成可能性予測。エネルギーでは不十分だった合成可能性予測を深層学習で行う。合成不可のラベル付けは本来困難であるが、本手法では文献に登場回数が多く、かつ報告されていない結晶構造を合成不可とラベル付けしたことで精度が高いモデルの構築に成功。

論文リンク
https://www.nature.com/articles/s43246-021-00219-x
著者/所属機関
Ali Davariashtiyani, Zahra Kadkhodaie & Sara Kadkhodaei
投稿日付
2021/11/18
詳細
-
文献に出てくる組成top108件に対し、確認されていない構造を異常結晶とする。数のバランスを取るために、多くは生成しない。(既存結晶の数程度までにとどめる)→合計600件
-
正例は3000件。このうち368件はtop108の組成の構造。他はランダムに抽出。
-
より多くの組成で異常結晶を加えて学習すると精度が低下した。おそらくは十分に調べられていない系に対し未観測のデータに負例をつけるのはラベル間違いのリスクがあるため。
-
モデルは3次元のボクセルに構造を押し込んでいる。解像度が悪い(0.54Å)ので原子間距離が合成可能性に重要な場合はモデルのアーキテクチャを変えることでより良い精度が出ると思われる。(単純に分割数を多くしても良いが)
-
e_above_hullは合成可能性の予測によく使われるが、実際はあまり機能しない。
-
amorphous solidが過去に提唱されたが、これは合成できる上限を示すもので、e_above_hullが小さいが合成できないものを予測できるわけではない。また、組成ごとに計算をしなければならない欠点があった。
-
電極材料で性能を実証
-
入力はボクセルごとに3チャネルあり、それぞれ原子番号、周期、族が正規化されたものが入力される。
疑問点
- 異常結晶はテンプレートベースで作られているが、構造緩和したのか?
- エネルギーに基づいて異常結晶を作成できないのか?異常結晶を多くすると精度が下がったのはどういうサンプリングをしたのか分からない。
- 学習データの組成分布が分からない。
参考文献
- Tang, B. et al. Machine learning-guided synthesis of advanced inorganic materials. Mater. Today 41, 72–80 (2020).
合成条件を機械学習で最適化 - Sun, W. et al. The thermodynamic scale of inorganic crystalline metastability. Sci. Adv. 2, e1600225 (2016).
e_above_hullが合成可能性指標として機能しないことを示した。
[2022] MDM: Molecular Diffusion Model for 3D Molecule Generation
概要
拡散モデルを用いた分子の3D構造生成。原子距離が近いものと遠いものを別個の同変性ネットワークで取り扱い、さらにネットワーク内にVAEを通して得たノイズ項を導入することで、高精度かつ多様性のある構造生成を実現。SOTAを大きく更新。
論文リンク
https://arxiv.org/abs/2209.05710
著者/所属機関
Lei Huang,*1 2 Hengtong Zhang,† 1 Tingyang Xu,1 Ka-Chun Wong 2
1Tencent AI Lab, 2City University of Hong Kong
投稿日付
2022/9/13
背景
-
分子生成で自己回帰モデルやflowベースの手法が提案されていたが、初期で大きく外れる構造が出ると、誤差が蓄積されうまくいかないという課題がある。
-
拡散モデルベースの分子構造生成では、大きく性能が向上したが、特に大きな分子において性能が低く、多様性も乏しかった。これは以下の2点が原因と考えられる。
- 3D構造生成ではデータは点群として扱われ、隣接原子との間の制約を明示的に扱うことが難しい
- 拡散モデルはランジュバンダイナミクスを用いたデノイジングスコアマッチングに相当するため、原子はデータ分布の勾配に沿って移動し、固定された初期ノイズにおいては似たような軌跡を辿ることが多い
新規性
- 上記の問題解決に取り組んだMDM(Molecular Diffusion Model)を提案。
- 異なる2つの結合タイプを考え、それぞれ同変性のエンコーダを用いて明示的に取り扱う。
- 構造の多様性を上げるため、データ分布をコントロールする潜在変数を導入。
手法
MDM概要
-
2つの原子がある閾値以下であれば共有結合(local edge)、閾値以上であれば結合はなく(global edge)、ファンデルワールス力がメインであるとして扱う。これにより隣接原子ペアの結合のようなものを扱うことができる。これを2つの同変なエンコーダで取り扱う。
-
VAE型のノイズ生成器を導入。これにより決定的な出力を避け、生成構造の多様性を確保する。VAE部分を学習することで、データ分布に適合するノイズを出力するようになるということ?

(sがガウスノイズの平均を算出するためのパラメータ)
- 分子のジオメトリは回転、並進に対し不変であるため、データ分布もそれらの処理に対し不変である必要がある。これはマルコフカーネルが同変なニューラルネットワークでパラメータ化されていると達成することができるため、同変なネットワークを設計した。

同変なマルコフカーネル
-
2Å以下のエッジはlocal edgeとした。local edgeとglobal edgeは別々のエンコーダに流れ、それぞれ化学結合間の力、ファンデルワールス力をモデリングする。
-
roto-translationに対し原子特徴量は不変、原子位置は同変である必要がある。Edge MLPでエッジ特徴と距離の特徴を変換し、SchNetのアーキテクチャに流す。これでノード特徴量が得られる。さらに1層のNode MLPを問い押し、原子の特徴量潜在空間を得る。SchNetは距離しか用いていないのでネットワークはroto-translationに対し不変。
-
原子位置のエンコードは以下で行う。

sθ(dij)はノード特徴とエッジ特徴を変換したもの。sθ(d)は不変操作であるが、ri-rjが同変であるので全体として同変操作になる。
ノイズ
逆拡散の過程は以下で行う。

学習

サンプリング

2つの同変なネットワークがノイズのパラメータを持つことで多様性が生まれる。
結果
- MDMはSOTAを大きく更新

MDM-NVはネットワークのノイズ部分を無くしたもの。MDMはMDM-NVに比べてuniquenessとnoveltyが向上しており(GEOMでは下がってるが)、可変ノイズを加えることで多様性が向上した。
参考文献
[2022] Supervised Pretraining for Molecular Force Fields and Properties Prediction
概要
分子の力場や物性予測の下流タスクは、関連の深いエネルギー予測を事前学習することで性能が大きく向上することを示した論文。事前学習のデータセットが安定構造であれば、forceのL1ノルム正則化を用いるとさらに効果的。事前学習によりモデルは分子の構造を理解するようになっており、幅広い下流タスクでの性能向上が可能。
論文リンク
https://openreview.net/forum?id=OFRe3QiPCM2
著者/所属機関
Xiang Gao, Weihao Gao, Wenzhi Xiao, Zhirui Wang, Chong Wang∗, Liang Xiang ByteDance Inc.
投稿日付
2022/9/27
背景
分子の物性データは、実験や重い量子計算をする必要があり量が少ない。事前学習が有効なアプローチであり、これまでは自己教師あり学習による事前学習が行われてきた。基本的な物理量に関する事前学習を行うことで、下流の物性予測タスクの精度向上が期待できる。
新規性
分子のエネルギーを予測するじぜんがくしゅうを行い、下流タスクの性能を評価。エネルギーを事前学習のターゲットとして選定した理由は以下。
- データ数が多い
- ノイズが少ない。エネルギーは量子化学計算で得ることができ、実験データと比べてノイズが少ない。
- 下流タスクのターゲットに関連する量。分子の物性は分子間や環境の相互作用により決定される。これはforceが関わっており、必然的にエネルギーも関わっていることになる。
以下のような下流タスクで大きな性能向上が確認された。

手法
下流タスクとして力場の予測、物性の予測の2系統を行った。
事前学習
PubChem PM6データセットを用いて事前学習を実施。8600万件の安定分子のエネルギーのデータセット。これは小分子で構成されるので、下流タスクが大きな分子である場合には向いていない。
エネルギーのMAEをロス関数として事前学習を実施。また、安定状態のデータであるため、force(エネルギーの微分値)は0に近いはずであり、制約項としてforceのL1ノルムを加えた。
また、分子の物性予測は入力がSMILESであることがよくある。そのような場合には事前学習として、SMILESから予測される原子位置を入力としてエネルギーを予測し、誤差をロスにする。このとき座標は正確ではなく、forceも不正確であるためforceの制約項は導入しない。
fine-tuning
最終層はランダムに初期化。その他は事前学習済みの重みを使用。
モデル
モデルは力場予測ではGemNet-T、物性予測ではEGNNを使用。
結果
力場予測
in-domain(同じ分子で座標が異なるテストデータ)に対する結果は以下。

事前学習により精度が向上している。
また、10分子のデータに対し、9分子で学習し、残りの1分子を予測するout-of-domainのタスクを行った結果が以下。

事前学習を行うことで精度が大幅に改善した。このとき、事前学習においてforceの制約項を入れることで性能がさらに向上することが分かった。
ベンゼンとトルエンは比較的小さいエラーであった。これはベンゼン環が含まれる分子が学習データ中に多いことから、in-domainっぽくなったためであると考えられる。substructureが学習データに含まれると、out-of-domainでもある程度汎化することが分かる。
物性予測
物性予測の結果は以下。

事前学習モデルが多くのタスクで最高性能であった。また、スクラッチで学習する場合より収束が大幅に速くなった(300 →30 epoch)。さらに、全てのタスクにおいて事前学習により精度が一貫して向上しており、事前学習は多くの場合で有効であることが分かった。
モデルは事前学習で何を学んだか
モデルのノード特徴量を取り出し、1層のLinear layerで元素種、原子座標を再構築するタスクを行った。このとき最後のLinear層のみ学習しGNN部分は固定した。

GNN部分をランダムにした場合と比較して、事前学習済みのモデルでは元素種や原子座標を再構築する精度が高かった。これは事前学習により構造情報を学習していることを示唆する。
また、どの層のノード特徴量を取得するかで結果が大きく異なる。元素情報は1層目が最も情報を含んでおり、ランダム初期値のモデルでは、取得する層を下流にするにつれてエラーが急激に大きくなる。これは元素情報が失われていることを示す。一方で事前学習したモデルは下層でもそれほどエラーが大きくならず、元素情報が下流まで伝わっていることが分かる。
分子のsubstructure予測でも同様の傾向が見られた。下層にいくにつれてエラーが小さくなり、グローバルな情報を学習できていることが示唆された。

参考文献
[2021] Human-in-the-loop for a Disconnection Aware Retrosynthesis
概要
single-stepでの逆合成解析において、切断部位をユーザーが指定可能なモデルを提案。切断される結合に関与する原子のみタグづけされたSMILESを入力とする。今回はRXNMapperで自動で切断部位を検出。テストデータにおいて88.9%の確率で正しい切断部位を予測できた。

論文リンク
https://openreview.net/forum?id=-xfwlkmsfN1
著者/所属機関
Andrea Byekwaso, Philippe Schwaller, Alain C. Vaucher, Alessandra Toniato, Teodoro Laino
投稿日付
2021/11/16
背景
従来のsingle-stepの逆合成予測モデルでは切断部位を指定できなかった。訓練データに依存した前駆体が提案されるので化学者が望む切断部位に基づく前駆体が必ずしも得られない。
新規性
切断部位を指定できるモデルを構築
手法
-
transformerを用いて翻訳モデルとして学習。入力のSMILESには、切断部位の原子に「1」のタグをつける。
-
データセットの構築はRXNMapperを用いる。反応により結合原子が変化した原子を検出。検出された原子に「1」のタグをつける。その他のタグは消去。
結果
- 14.1%の反応が完全に正解と一致した。
- 88.9%の反応は切断部位を正確に予測できていた。
参考文献
[2022] GemNet-OC: Developing Graph Neural Networks for Large and Diverse Molecular Simulation Datasets
概要
巨大なデータセット学習用に最適化されたGemNet-OCを提案。OC20でSOTAを記録。様々なデータセットでこのモデルの最適パラメータを調査。データセットの複雑性により最適値が大きく異なり、一貫性を持たせるためには複雑性が同一程度で、かつ巨大なデータセットである必要があることがわかった。これは巨大なデータセットの均一サンプリングでも実現でき、OC-2Mが計算コストを抑えながらモデルのベンチマークが可能である。今後のモデルの発展のために、いくつかのモデルでOC-2Mのベンチマークを作成した。
論文リンク
https://openreview.net/forum?id=u8tvSxm4Bs
著者/所属機関
投稿日付
2022/7/7
背景
近年のデータセットは以下の特徴がある
- 元素数が多い
- システムサイズ(原子数)が大きい
- データセットサイズが大きい
- 学習データとテストデータのドメインシフトが大きい
これらの違いがあるにもかかわらず、GNNのベンチマークは昔ながらの小さいデータセットで行われている。小さなデータセットを用いたときのモデルの精度向上は、大きなデータセットを用いたときにも当てはまるかわからない。極めて小さいデータセットでは、単純なモデルがSOTAモデルより高精度である例もある。
データセットの例は以下

OC20以外のほとんどのデータセットはテストデータが学習データと同じトラジェクトリから取得されており、OODの評価ができない問題もある。
新規性
巨大なOC20データセットに基づいてGemNet-OCを開発。さらに複数の異なるデータセットで性能を検証し一貫性があるかを調べた。
手法
データセット
OC20は分割したサブセットを作成。①触媒の元素をRb, Snに限定したOC-Rb, OC-Sn、②30原子以下の構造に限定したOC-sub30、③ランダムに一部を取得したOC-200k, OC-2M、④30原子以下かつRbに限定したOC-XSを作成。
これらはそれぞれ、①化学的多様性、②原子数多様性、③データサイズ多様性、④化学的・原子数多様性の複合、の影響を調査するために作成。
GemNet-OC
GemNetを元に、OC20に最適化。GemNet-OCではmessage passingにノードとエッジのembefddingを渡し、それぞれアップデートする。このとき隣接原子のノード、エッジ特徴および構造的情報(距離、角度、二面角)を用いる。
モデルの全体像は以下。GemNetからの変更点はオレンジで示している。

隣接原子の取得
通常、カットオフ内の原子を隣接原子とするが、OC20のような化学的多様性が高いデータセットでは、ほとんどの原子間が6Å以上離れているケースや、全てが3Å以下であるケースなど様々である。このため、単一のカットオフを使用すると、エネルギー予測が困難な切断されたグラフができたり、過剰なエッジ数で計算コストが高いグラフができたりする。
これに対処するため、固定された数の隣接原子を使用した。これは、ある小さな変位によって2つの原子の順序が入れ替わる場合は、エネルギーの微分ができなくなり、好ましくないと思われたが、実際にはそれほど問題にならなかったとのこと。
これにより、精度が高くなり、スループットが3倍になった。
基底関数
GemNetでは原子間距離を球ベッセル関数、角度を球面調和関数で展開していたが、計算負荷が高かった。これらを単純化し、精度の低下なしに29%の高速化を実現した。
4体間相互作用の取り扱い
4体間相互作用の計算は非常に重いので、近い隣接原子間のみに限定した(遠くなると急速に減衰することを確かめたそう)。
これにより、元の実装では330%のオーバーヘッドがあったのを、わずか31%に抑えることができた。
この相互作用はノードのembedingに加えられる。
その他の変更
- アウトプットをembeddingにして、MLPを接合し表現力をあげた。
- 原子のembeddingにMLPの出力を都度加算
- 原子のembeddingをエネルギー出力層に加算し、出力の情報が原子のembeddingにより伝わるようにした
結果
OC20データセットでの、ベンチマークモデルとGemNet-OCの精度比較は以下。

GeNet-OCが最も精度が高かった。
さらにGemNet-OCはGemNet-dTの精度を600 GPU時間で超え、学習時間も短くできた。パラメータを大きくしたGemNet-OC-Lでも依然として学習時間が短くできた。
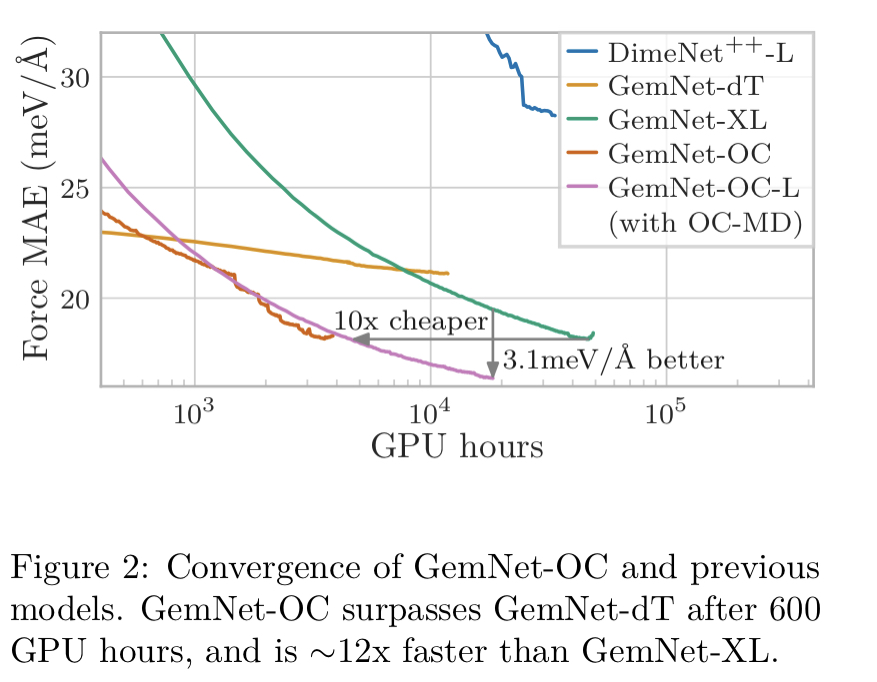
このように、データセットに合わせてモデルを最適化することで精度を上げられることが可能であることがわかった。
モデルの幅と深さ
モデルの幅や深さはデータセットごとに異なる最適値を示した。データ数が小さいほど幅が狭いネットワークが最適であった。

特に、モデルサイズを大きくしてもOODデータに対してはほとんど改善は見られなかった。この結果は、モデルのスケーリングだけでは、分子の機械学習はうまくいかないことを示唆している。
バッチサイズは32-128程度が最適な場合が多かった。MD17では少ないバッチ数による汎化効果も見られた。
エッジのパラメータ

4体間は遠くまで拾ってもあまり効果なし。
エッジの距離を展開する関数は精度に大きく寄与する。
その他の効果

GemNetで提案されたscaling factorは初期の学習安定化には効果的であるが、最終の精度にはそれほど影響を与えない
参考文献
[2022] TeaNet: Universal neural network interatomic potential inspired by iterative electronic relaxations
概要
EAMやTersoffポテンシャルの情報の流れを模倣し、テンソル量をモデルに組み込んだTeaNetを提案。原子と結合にそれぞれ特徴量を持たせ、繰り返し更新しており、EAMの一般化であるとみなせる。角度情報は、生の値を入力するのではなく、テンソル積により表現している。層を重ねることはエネルギー最適化の反復を模擬していることになる。様々な元素かつ大きく歪んだデータセットに対し高精度に予測が可能であった。
論文リンク
https://doi.org/10.1016/j.commatsci.2022.111280
著者/所属機関
So Takamoto, Satoshi Izumi, Ju Li
投稿日付
2022/3/9
背景
これまで機械学習ポテンシャル(NNP)はスカラーやベクトルのみを扱うものが多かった
新規性
DFTが収束するときのデータフローを考えると、テンソル量を扱うことで精度が向上すると期待できる。それを組み込んだTeaNetを開発。
EAMの古典ポテンシャルはTeaNetの派生系であると見なすことができる。
手法
活性化関数
活性化関数としてsoftplusを積分したものを使う。力はエネルギーの微分値として得られ、それをロス関数に加えるため、2階微分が発生する。このときの関数形がsoftplusになるように活性化関数を設定した。
preprocess
preprocess/postprocessの概要は以下

原子特徴量はスカラー部分、ベクトル部分、テンソル部分があり、結合特徴量はスカラー部分、ベクトル部分がある。これを更新していく。
原子特徴量のスカラー部分は電子軌道の占有を模擬したone-hot-likeな特徴量で初期化。ベクトル部分、テンソル部分はゼロで初期化した。
結合の特徴量として、スカラー部分は
で初期化する。モースポテンシャルを模擬している。ベクトル部分は0で初期化。
postprocess
postprocesでは、更新された原子特徴量、結合特徴量をそれぞれ線形変換し、足し合わせることでエネルギーを得る。
local interaction block
まず、原子特徴量と結合特徴量それぞれに対しMLPで特徴量を変換する。

このとき、ベクトル部分、テンソル部分の取り扱いはスカラーの乗算、内積、テンソル積で行わないといけない。今回はベクトル部分をスカラーにしている。

これらを用いて結合ごとの特徴量を作成する。結合に関与する原子特徴量、結合特徴量を入力にする。
まず、原子特徴量のテンソル部分と結合ベクトルの内積をとり、ベクトル部分に合算する(おそらくi, jに対しconcatした特徴量で行う)。
この、結合の原子由来の特徴量(β)と結合由来の特徴量の相互作用を以下で計算する。

ここでの出力はベクトル部分、テンソル部分の特徴量は全てスカラー特徴に圧縮されている。
また、カットオフ以上での値が0になるように、それぞれの特徴量にカットオフ関数を適用している。
このようにして得られた特徴量群を以下のようにして集約する。

さらに、集約した特徴量を以下のようにして、原子部分、結合部分の特徴量に再び変換する。

この特徴量を原子のneighborで集約して元の特徴量のskip-connectionを加えることで、更新された特徴量を得る。
データの更新がやや複雑だが、原子、結合にそれぞれ高次元特徴量(ベクトル、テンソル)を持たせ、さらに結合ベクトルも加えた全特徴量に対し、うまく相互作用を計算しアップデートするというGNNになっている。勾配の伝達をサポートするためskip-connectionが入っている。
古典ポテンシャルとTeaNetの関係
EAMポテンシャルは以下のように、1層のGNNと考えることができる。

原子情報は対応する結合に分配され、結合ごとの特徴が計算される。これの一部を原子情報に流入させ、原子情報をアップデートし、最終的に結合情報と統合しエネルギーを得る。TeaNetはこの情報の流れを模倣している。
TeaNetは結合ベクトルの情報は距離のみに制限している。明示的に角度情報を入れ込むモデルもあるが、角度はノードでもエッジでもなく3体間の組み合わせの特徴量であり、ノード特徴量に落とし込むには、対称関数などのアドホックな関数が必要になってしまう(表現力が落ちることを言いたい?)。
角度情報はノードとエッジのベクトルとテンソルを用いた畳み込み操作で表現できるとのこと。Tersoffポテンシャルを書き直すと以下の形になる。

全てのΣはiにしか依存していないため、畳み込み操作で計算できる。これにはテンソル部分が必要になり、他のモデルでは球面調和関数により導入されているが、TeaNetでは純粋なテンソル量として取り入れる。
学習
高温のMD計算で大きく歪ませた構造に対し学習を実施。
層を重ねると不安定になったので、学習初期は全ての層のパラメータを同一にして行った。
結果
層を増やしても過学習が起こらず精度が向上した。また、ablation studyでは、テンソル量導入、ゲート関数(原子ごとの特徴量変換)、活性化関数の変更が有効であったことが示された。

参考文献
[2022] NeuralNEB – Neural Networks can find Reaction Paths Fast
概要
遷移状態のデータセットで学習した機械学習ポテンシャルモデルを用いてNEB計算することで高精度に活性化エネルギーを予測。DFTと比較して1500倍の高速化を実現。QM9などの平衡状態のデータでは全く予測ができておらず、NEB計算時のデータセットの重要性を示した。
論文リンク
https://arxiv.org/abs/2207.09971
著者/所属機関
Mathias Schreiner, Arghya Bhowmik, Tejs Vegge, Ole Winther Technical University of Denmark (DTU), 2800 Lyngby, Denmark
投稿日付
2022/7/21
背景
遷移状態のデータが不足しているため、NEBなどのシミュレーションを機械学習ポテンシャルで行うことは難しい。
新規性
PaiNNとTransition1x(反応パス中の分子構造のデータセット。1000万件)を用いてNEBの精度を検証。
手法
NEB
- NEBの初期配置はIDPP(非常に計算コストが低いポテンシャル)により作成。パスの最適化にはBFGSを用いた。
データ
-
ANI1x, QM9x, Transition1xのデータセットでそれぞれ学習させた。ちなみにこれらのデータセットの基底関数は同じなので組み合わせることも可能
-
ANI1xは平衡状態ではない分子配置を計算している。配置は、分散が大きくなるように能動学習で収集されている。全部で500万件のデータセット。
-
Transition1xはMDでは収集が不十分になってしまう遷移状態の配置を、DFTでのNEB計算により収集している。全部で1000万件のデータセット。
-
QM9は13万件の分子のデータセット。全て平衡状態のデータ。本論文ではこのデータセットを6-31G(d)、ωB97xで再計算し、ANI1x, Transitionxと計算条件を合わせたQM9xデータセットを作成。条件が変わったのでQM9xは平衡状態である保証はない。
モデル
-
PaiNNはテンソル量を予測するために回転同変性を備えたGNNモデル。本論文ではカットオフ半径を5Å、256ユニットの中間層のmassage passing layerを3つ配置したモデルで学習。
-
学習時、Transition1xは反応ごと、ANI1xは組成ごと、QM9xはランダムに分割した。
結果
- それぞれのデータセットで学習し、DFTでのNEBと比較した結果は以下の通り。

QM9xで学習したデータは著しく精度が悪く、平衡状態の配置から遷移状態を予測することができないことを示している。
Transition1xは最も高精度であり、DFTの1500倍速く、かつDFTに近い結果が得られた。
- Transition1xで学習したモデルでPESを評価した結果は以下の通り。

モデルは活性化エネルギーを正しく予測できるだけでなく、DFTのPESをうまく再現できている。
- それぞれのモデルとDFTの計算値の関係は以下の通り

Transition1xは概ね正しく予測できている。QM9xは全く予測できていないが、ANI1xは系統的な誤差がある。ANI1xは外挿領域のエネルギーを高く予測する傾向にあった。ちなみに、この系統的な誤差を補正してもなお、Transition1xの方が精度が高かった。
参考文献
[2022] EQUIVARIANT 3D-CONDITIONAL DIFFUSION MODELS FOR MOLECULAR LINKER DESIGN
概要
分子のリンカーを拡散モデルで予測するDiffLinkerを提案。これまでのモデルと異なり、任意の数のフラグメントを取り扱える。また、結合する原子やリンカーの原子数を指定する必要は無く、モデルが自動で判別する。ノイズの予測にはEGNNをベースにしたE(3)同変なネットワークを使用。フラグメントやタンパク質ポケットの点群データで条件付けすることが可能。複数のデータセットで、他のモデルより高い精度を示した。
論文リンク
https://arxiv.org/abs/2210.05274
著者/所属機関
Igashov, Ilia and Stärk, Hannes and Vignac, Clément and Satorras, Victor Garcia and Frossard, Pascal and Welling, Max and Bronstein, Michael and Correia, Bruno
投稿日付
2022/10/11
背景
- タンパク質のリンカーの設計はSMILESを用いた手法や自己回帰モデルによる手法が提案されていたが、原子入れ替えについて同変でなく、フラグメント2つを結合させることしかできなかった(複数の結合は不可)。
- リンカー設計はタンパク質ポケットに依存するため、その情報を適切に使う必要があるが、これまでのモデルはできていなかった。
新規性
-
リンカーを生成する条件付き拡散モデルDiffLinkerを設計。元素種と原子位置をサンプリングし、入力のフラグメントで条件付けられたNNでそれらをアップデートしていく。
-
DiffLinkerは以下の特性がある。
- 並進、回転、鏡映、入れ替えに対し同変である
- 入力のフラグメント数に制限がない
- リンカーに関する事前情報が不要
- 取り囲んでいるプロテインポケットの情報を渡すことができる
手法
- 全体像は以下

-
分子は点群データとして扱う。原子位置と元素種のone-hotベクトルを結合して原子の特徴量とする。元素は離散値であるため、one-hotにガウスノイズを加え、連続値っぽくしておく。更新後の元素判別はargmaxをとることで行う。
-
リンカーの点群データzはフラグメント(オプションでタンパク質ポケットも)の点群データuで条件付けられる。このuはプロセス中変化しない。

-
ノイズを算出する変換関数およびuを変換する関数が同変であればp(z0|u)も同変になる。uの変換は今回は「どの原子がリンカーと接続しやすいか(アンカーはどれか)」という情報にした。具体的にはアンカーの質量中心?と定められた。現実的にはアンカーが不明なことが多いので、u全体の質量中心を用いても良い。
-
モデルのダイナミクスを決める関数φはE(3)同変なEGNNを修正したものを用いた。リンかーの情報ztとuを受け取り、1つのグラフとして扱う。ノード特徴量は原子特徴量、原子位置、時刻、フラグメントのフラグ、アンカーのフラグで構成される。出力はノイズで、座標情報と原子情報のノイズが結合されている。φをinvariantにするため、座標は変換前の値を引くらしい。

EGNNのレイヤーではノード間の距離、ノード特徴をNNで変換し、メッセージを算出。rのアップデートはリンカーのみ行い、フラグメント(u)には0を割り当てる。φe, φhはスカラーのみなのでinvariant、φvelは座標に関して線形なのでequivariantになり、全体としてequivariantな変換になる。
-
フラグメント間のリンカーサイズを予測するために、フラグメントのグラフを作成し、GNNでリンカーサイズを予測するモデルを別途用意した。
-
uはプロテインポケットの情報も取り入れることが可能。
結果
モデルのベンチマーク結果は以下。


特に、生成された構造の妥当性が他のモデルより高かった。自己回帰モデルでは生成ステップの各時刻において価電子の妥当性を担保するルールを明示的に組み込んでいる。DiffLinkerではそれを組み込んでいないが、データから学習できている。
- 他のモデルでは2つのフラグメント間しか考慮できないが、DiffLinkerでは任意の数のフラグメント間のリンカーを一気に生成できる。
参考文献
[2022] Deep Learning and Symbolic Regression for Discovering Parametric Equations
概要
NNの活性化関数をprimitive function(sin()など)に置き換え、シンボリック回帰を行うEQL networkを改良し、任意の時刻変数での予測を可能にした。L0正則化を用いることで重み(係数)がスパースになり解釈性が高い式が得られる。偏微分方程式等で高い精度が得られ、外挿も可能であることを示した。
論文リンク
https://openreview.net/forum?id=zjbSx8-m1wJ
著者/所属機関
Samuel Kim, Michael Zhang, Peter Y Lu, Marin Soljacic
投稿日付
2022/5/21
背景
-
深層学習は内部がブラックボックスであり、サイエンスの分野において、モデルから重要な知見を導き出すことは難しい。シンボリック回帰はデータに適合する数学的な式を機械学習により導くことができ、解釈性の高いモデルを与えるが、複雑な系に適用するのは難しかった。
-
ニューラルネットワークの活性化関数をprimitive functionに変更することでシンボリック回帰を行うことで、複雑な系にも対応できることが知られている。
新規性
新しいアーキテクチャを提案。時間に応じた予測が可能になる。(時間により係数が変化するが、根底の式は保つようにする)
手法
EQLネットワーク
EQLネットワークは活性化関数をprimitive functionに置き換えたニューラルネットワークのアーキテクチャ。

活性化関数がsin()や乗算などに変更されている。つまり通常の線形変換の後にそれぞれのユニットに対しprimitive functionを適用する。複数のユニットからの入力も受け付ける(乗算等の場合)。次の層ではこれら出力の線形和になるので、複雑な式が表現できる。学習は通常のNNと同様に行い、それぞれの重み(式の係数)が決定される。
これは既に報告されており、本論文では以下のいくつかの変更を行った。
- 解釈性の高いモデルを得るため、モデルの重みはスパースにする必要がある。本論文ではL0正則化を用いた。L0正則化では微分不可能になるため以下のようにゲートzを定義し、微分を可能にしている。

-
単純な関数にも対応させるため、スキップ接続を追加。(序盤の層の出力は単純なので、それだけで表現できるものに対応するため)
-
重みWを時間の関数W(t)に変更した。時間を入力に受け取り層の重みを出力する。これにより時間に応じた重み(式)を学習できる。これに上記のゲート関数zを適用したのち、primitive functionを適用する。

このアーキテクチャにより、連続的なドメイン(時間など)での予測が可能になり、異なる時間でのデータを扱うことができる。
学習
それぞれの時刻に対し512個の学習データを作成。時刻は128種類用いた。合計で65,536件のデータで学習。さらに学習範囲外の入力に対してテストを行った。
結果
- 学習結果は以下

時間に応じた式を学習できていることが分かる。また、以下の図(上)ように、外挿領域(赤色部分)についても一致していることが分かる。図(下)のように係数も一致

これらの式はsgnやsin内にtが入っており、線形回帰のテクニックでは見つけられないが、本手法では関数がネストされているので見つけられるとのこと。
- 偏微分方程式も精度高く学習できる(今回は移流拡散方程式)。

- 入力を潜在変数に変換してからモデルに渡すこともできる。(よくわからなかった)

-
本モデルは、例えば一部分が解析的に分かっているが、残りが分からない系に対して適用し分析することが有用である。
-
モデルが完全に収束しない場合は、パラメータを抜き出し、ファインチューニングをすることで、より正確な予測が可能になる。
参考文献
[2022] Learned Force Fields Are Ready For Ground State Catalyst Discovery
概要
構造緩和において、機械学習ポテンシャル(NNP)での緩和はDFTよりも安定な構造に収束する確率が高いことを示した。これはNNPはforceにノイズが含まれており、局所解を抜け出せるため。また、NNPでは最適化アルゴリズムをAdamWなどのモーメントを使用するものに簡単に変えることができ、これも局所解を抜け出すことに有利。また、調和ポテンシャルのみで学習したモデルでもDFTよりも安定構造を見つける可能性が高いことが判明。これはエネルギー局面が滑らかになり、局所解にトラップされにくいため。触媒の安定構造の発見において、既に機械学習ポテンシャルはDFTを代替可能なところまできているとのこと。
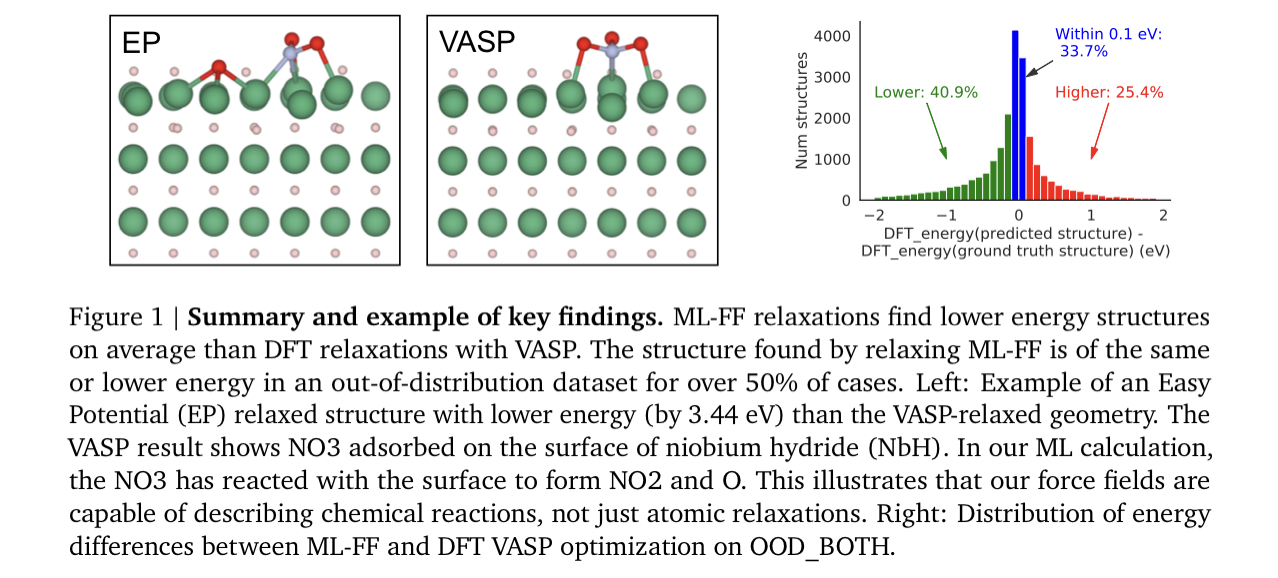
論文リンク
https://arxiv.org/abs/2209.12466v1
著者/所属機関
Deepmind
Schaarschmidt, Michael and Riviere, Morgane and Ganose, Alex M. and Spencer, James S. and Gaunt, Alexander L. and Kirkpatrick, James and Axelrod, Simon and Battaglia, Peter W. and Godwin, Jonathan
投稿日付
2022/9/26
背景
- DFT計算による構造緩和は計算コストが高く機械学習ポテンシャル(NNP)による代替が求められている。
新規性
NNPは多くの場合でDFTよりも安定な構造を見つけることができることを示した。これは、forceのエラーが大きくても成り立つ上、安定サイト周辺のみを学習した場合でも成り立つ。
手法
-
モデルはGraph Net Simulator(GNS)を使用。forceはenergyの微分で得るのではなく、最終のnode特徴量を変換して得た。この実装では、構造緩和時に使用しないenergyは不要であるが、検証のためenergyも出力した(同じくnode特徴量を変換し、合計を取った)。学習したモデルをML-FFと表すことにする。
-
構造緩和は一般的にforceを用いるが、PESを近似するサロゲートポテンシャルの勾配を用いることも可能である。PESよりもサロゲートポテンシャルがスムーズであれば、最適化が高速になり、局所解を抜け出す可能性が高くなる。特に放物線であれば最適化が簡単である。しかし、放物線近似は最適解が分かっている必要があるためOODのサンプルには適用不可である。しかし、機械学習で多くのサロゲートポテンシャルを学習することで、OODサンプルについても最適解を予測できるようになる。
これには調和ポテンシャルを使用した。初期構造と収束構造を内挿した原子位置に対しガウスノイズを加え、変位量に応じた調和ポテンシャルを作成(安定構造以外も含む?合ってる?)。これを学習したモデルをEasy-Potential(EP)と表すことにする。

-
構造緩和はAdamWを用いて行った。これはノイズが多いforceに対して適切なoptimizerであると考えられる。学習率は0.1に設定。forceの負値を勾配として最適化を実施(エネルギーの微分ではない)。EPでの緩和の場合はforceのエラーが大きく、緩和後の構造が不適切なため、ML-FFでさらに緩和した。
-
データセットはOC20を使用。
結果
- 50%以上のケースでML-FFはDFTよりも安定な構造に収束。OODサンプルについても同様。

また、DFTの収束構造よりも安定であった予測構造は、DFTの収束構造と原子位置が大きく異なっていた。これはML-FFは局所解を抜け出して、より安定な解に収束したことを示す。
- EPでの緩和はさらに良い結果が得られた。

また、ML-FFの緩和よりも少ない更新回数で収束し、高速に計算が可能であった。
- ML-FF使用時の、デバイスごとの計算時間の比較は以下。GPUで圧倒的に加速できる。

- ML-FFで緩和した時のtrajectoryに対し、forceをプロットした結果が以下。かなりノイズが多いことに加え、AdamWをoptimizerとして用いることにより、局所解を抜け出すことをサポートしている。

- 二体間ポテンシャルも、明示的に学習させてない割には多くの場合で良い一致を示した。

-
EPとVASPを組み合わせることで、88%の構造で、VASPのみの緩和よりも安定な構造が得られた。そのため、ML-FFが信用できない場合でも、EPによる緩和をVASPの前段階に加えることで、より安定な構造を高速に見つけることが可能である。
-
本論文の結果は、NNPの進展は緩和後のこうぞうの一致度や、forceの正確さのみで語られる必要はないということを暗示している。
参考文献
GNS:
A. Sanchez-Gonzalez, J. Godwin, T. Pfaff, R. Ying, J. Leskovec, and P. Battaglia. Learning to simulate complex physics with graph networks. In H. D. III and A. Singh, editors, Proceedings of the 37th International Conference on Machine Learning, volume 119 of Proceedings of Machine Learning Research, pages 8459–8468. PMLR, 13–18 Jul 2020. URL http://proceedings.mlr.press/ v119/sanchez-gonzalez20a.html
[2021] Large-Scale Representation Learning on Graphs via Bootstrapping
概要
グラフデータの表現学習手法(BGRL)を提案。BYOLをグラフ用にしたものでメモリ効率がよく、大規模なグラフに対しても適用可能。これまでの手法に比べ2-10倍のメモリ効率で、同等あるいは上回る精度を獲得。

論文リンク
https://arxiv.org/abs/2102.06514
著者/所属機関
Shantanu Thakoor, Corentin Tallec, Mohammad Gheshlaghi Azar, Mehdi Azabou, Eva L. Dyer, Rémi Munos, Petar Veličković, Michal Valko
- deepmind
投稿日付
2021/11/4
背景
- これまでのグラフ表現学習のSOTAは対照学習であり、ノード数の2乗の計算量とメモリを必要としていた。これは特に大きなグラフに適用する際に問題になる。
新規性
- Bootstrapped Graph Latents (BGRL)により、スケーラブルで精度の高い表現学習手法を提案。BYOLをグラフ用にしたもの。
手法
-
online encoderとtarget encoderを用いてノードを2種類にエンコードする。online encoderはtarget encoderの表現を予測することで学習され、target encoderはonline networkの指数移動平均によって更新される。これにより表現の崩壊(エンコーダーが全て同じ出力を返す)ことを防ぐ。2つのエンコーダーの出力は、似たようになりながらも異なってほしいという制約になる。対照学習のようにnegative sampleを必要とせず、大きなグラフにも容易にスケール可能。
-
BYOLと異なり、predictor networkを使わない。画像と違い、グラフのembeddingサイズは小さいため不要。
-
データ拡張はノード特徴量マスクと、エッジマスクを使用
-
online encoderのパラメータは、online表現を変換したものとtarget表現のコサイン類似度で学習される。このとき、target encoderのパラメータ更新は行わない。
結果
-
これまでの手法に比べ2-10倍のメモリ効率で、同等あるいは上回る精度を獲得。

-
より複雑なデータ拡張(これはメモリを多く必要とする)を行っても精度はほぼ変わらなかった。そのためBGRLはシンプルなデータ拡張のみでよく、大きいグラフに適用する際に複雑なデータ拡張が使えない場合でも、精度を担保できる。
-
モデルにGATを用いた場合、他の事前学習手法(SimCLRをグラフ系に適用したもの)と比較して全層のattention weightsのエントロピーはBGRLで大きくなった。これはattentionがうまく機能していることを示している。
-
教師あり学習時に、バッチ内にラベル無しノードを加えて、補助タスクとして自己教師あり学習のロスを加えて学習させると、ラベル無しノードの割合が増えるにつれて精度と安定性が劇的に向上した。

参考文献
[2022] Learning Local Equivariant Representations for Large-Scale Atomistic Dynamics
概要
スケーラビリティと精度を両立した機械学習ポテンシャルモデルAllegroを提案。原子のエッジはスカラーとテンソル特徴量をもち、SE(3) or E(3)同変な変換を繰り返すことでmessage-passingを行わず高精度を実現。QM9やMD-17のSOTAを更新し、1億原子の大規模なシミュレーションが可能。
論文リンク
https://arxiv.org/abs/2204.05249
著者/所属機関
author={Musaelian, Albert and Batzner, Simon and Johansson, Anders and Sun, Lixin and Owen, Cameron J and Kornbluth, Mordechai and Kozinsky, Boris},
journal={arXiv preprint arXiv:2204.05249},
投稿日付
2022/4/11
背景
- GNNを用いたモデル(MPNNs)の情報伝達は表現力が高いが、スケール化しにくい。これは受容野内の隣接原子は原子数の3乗で増加していくため。一方でACSFなどの局所構造を特徴量化したモデル(local MLIPs)では大きな系に拡張できるが、精度が低い。
新規性
- スケーラビリティと精度を両立したAllegroを提案。E(3)同変な表現のテンソル積を繰り返すことでmessage-passingを行わず高精度を実現。
手法
-
エネルギーは各原子の和で表現。それぞれの原子のエネルギーはさらに一次関数で変換される。係数は学習可能なパラメータ。各原子のエネルギーは中心と隣接原子間で表現されるpairwiseエネルギーの和で表現している。forceは通常通りエネルギーの微分で得る。(エネルギー保存則のため)
-
原子間のエッジはinvariantな特徴量とequivariantな特徴量の組み合わせで表現される。これらのテンソル積によって表現が更新される。最終層のスカラー特徴量をMLPで変換することでエネルギーを得る。
-
まず原子のone-hot表現と動径基底関数を結合したものをMLPで変換することで、エッジの特徴量を得る。これを各レイヤーに流す。
-
equivariantの特徴量は、まず球面調和関数の線形和によって得られる。
-
各レイヤーでの処理は以下の通り。まずスカラー特徴量から、重みをMLPで算出し、隣接原子との関係(球面調和関数)の線形和をとり、中心原子の環境を取り込む。これとテンソル特徴量のテンソル積を得る。得られた特徴量のスカラー特徴料部分と元のスカラー特徴量とのテンソル積でスカラー表現を更新。equivariantな変換でテンソル特徴量を変換。

-
density trickというもので計算コストを減らしているらしい。
-
SOAPなどとの違いは、隣接原子との球面調和関数の和の重みが学習可能であること。これにより後半の層では単純な2体間の情報よりもリッチな情報が取得できている。
結果
-
QM9とMD-17のSOTAを更新。
-
外挿領域のデータに対する汎化性能も良好(300KでのMDデータを学習し、600K, 1200Kのデータを予測)
-
Li3PO4のMDデータもよく再現した
-
16GPUの並列化で1億原子の超大規模なシミュレーション
参考文献
- 学習時のエネルギー正規化の方法
Sun, L., Batzner, S., Musaelian, A., Yu, X. & Kozinsky, B. On the normalization of potential energies for neural-network-based interatomic potentials training. In Preparation .
[2021] Learn2Hop: Learned Optimization on Rough Landscapes With Applications to Atomic Structural Optimization
概要
global optimizationの最適化アルゴリズムに関する論文。メタ最適化を取り入れ、原子の更新をNNで予測。緩和中に局所安定間のホッピングが発現し効率的に探索が可能。

これまでの問題設定での関数形状と本論文での関数形状

論文リンク
http://proceedings.mlr.press/v139/merchant21a.html
著者/所属機関
Amil Merchant, Luke Metz, Samuel S Schoenholz, Ekin D Cubuk Proceedings of the 38th International Conference on Machine Learning, PMLR 139:7643-7653, 2021.
google research
投稿日付
2021/6/18
背景
-
多峰性の関数において、global minimumを探索することは、ランダムな初期値をもちいた試行が多く必要であった。
-
たった147原子でもエネルギー曲面は10^60個の異なる局所安定が存在する
-
大域最適化は局所安定間の障壁が大きいとさらに困難になる。
-
これまでは多くの試行回数、よく設計されたハンドクラフトな特徴量、学習率の細やかな調整が必要であった。
新規性
-
メタ学習の枠組みを適用。さまざまなロス曲面を最適化するアルゴリズムを学習
-
更新式はニューラルネットワークで学習された関数を用いる
手法
-
ポテンシャルはLennard-Jones, Gupta Clusters, Stillinger-Weberを使用。このようなポテンシャルでもエネルギー局面は多くの局所解を持った複雑な形状になる。
-
設定したポテンシャルを用いて原子に働く力を算出。これらを特徴量化した後にNNに入力し、更新位置を算出。緩和が収束するまで繰り返し、最終エネルギーを取得。これをロスとしてNNの学習に使用。NNのパラメータは最終構造のエネルギーが低くなるように最適化される。そのためこのNNを用いて最適化を行うと、安定な構造が得られるようになるので、optimizerとして使用できる
-
特徴量は原子位置、力、1次,2次モーメント、対称関数などを使用。
-
高い障壁の存在と、滅多に現れないパターンが存在することが課題となる。原子位置が近すぎるとスパイクが生じ、学習が不安定になる。
→meta-lossのクリッピングを適用し対処。 -
meta-lossの勾配でパラメータを更新するのではなく、GAでNNのパラメータを更新するモデルも検討
-
本手法で最適化された構造は局所安定であることを保証しないので、最適化後の構造はさらに勾配降下法である程度緩和し最終構造を得た。
結果
-
学習されたoptimizerは既存の最適化アルゴリズム(FIRE,Adam)の性能を凌駕し、さらに汎化能力もあった。

点線は学習に使用した原子数だが、それを超える原子数でも性能が高い。 -
エネルギー分布も大幅に改善

-
学習されたoptimizerでは、緩和中に更新関数が大きくなり、局所安定間をホッピングしている挙動が確認された。
-
ablation studyでは、SINE特徴量と対称関数が影響が大きいことがわかった。SINE特徴量は最適化ステップを複数の周期のsin波でエンコードしたものであり、探索と活用を調整していると考えられる。
-
合金系において、違う元素系で学習したoptimizerでもうまく働くことを確認。(AgAu, AgPt, PdAuで学習したoptimizerでPdPtの最適化を実施)
参考文献
[2021] Deep learning for visualization and novelty detection in large X-ray diffraction datasets
概要
VAEによりXRDから構造の潜在空間を学習。潜在空間は構造の類似度や大まかなXRDパターンを認識できており、未知層や混合物に対し再構築誤差が大きくなり検出可能。これまでの分類モデルと異なり、未知層の検出が得意。

論文リンク
https://www.nature.com/articles/s41524-021-00575-9
著者/所属機関
Lars Banko, Phillip M. Maffettone, Dennis Naujoks, Daniel Olds & Alfred Ludwig
投稿日付(yyyy/MM/dd)
2021/11/9
背景
-
機械学習による相分類の手法が提案されてきたが、未知相に対しての予測精度が低かった。
-
本論文では実験やデータ分析のサポートとして、未知相の検出に焦点を当て、VAEを構築。
新規性
- VAEの再構築誤差を未知相
手法
-
XRDを入力としたVAE
-
組成も加え、conditional VAEとすることも可能
-
学習データはピークシフトなどの実験でよくあるパターンのデータ拡張されたXRDパターン
結果
-
潜在空間は空間群により綺麗に分類され、物理的に意味のある表現学習ができている。
-
既知相の再構築誤差は非常に小さく、未知相の再構築誤差は大きいため、再構築誤差により未知相の検出が可能。

- 混合物の再構築誤差が大きく、2成分の混合ではちょうど半量ずつの混合(学習データから最も遠いパターン)が最も再構築誤差が大きい。

参考文献
- XRDの自動同定
Maffettone, P. M. et al. Crystallography companion agent for high-throughput materials discovery. Nat. Comput. Sci. 1, 290–297 (2021).
[2022] BIGDML—Towards accurate quantum machine learning force fields for materials
概要
BIGDMLは長距離相互作用と対称性を取り込んだGPモデルにより少量のデータでPESを再現するモデル。GPの共分散行列計算時に、様々な対称操作で周辺化することで、対称性を取り込む。対称性を活用することで100以下程度のデータ数で高い精度、ロバスト性を獲得。GAP/SOAPと比較して10倍の精度を実現。
論文リンク
https://www.nature.com/articles/s41467-022-31093-x
著者/所属機関
@Article{sauceda2022bigdml,
title={BIGDML—Towards accurate quantum machine learning force fields for materials},
author={Sauceda, Huziel E and G{'a}lvez-Gonz{'a}lez, Luis E and Chmiela, Stefan and Paz-Borb{'o}n, Lauro Oliver and M{"u}ller, Klaus-Robert and Tkatchenko, Alexandre},
journal={Nature Communications},
volume={13},
number={1},
pages={1--16},
year={2022},
publisher={Nature Publishing Group}
}
投稿日付
2022/6/29
背景
これまでの機械学習ポテンシャル(MLFF)は局所的な特徴量がメインであり、長距離の相互作用をうまく取り込めていない。
より高度な計算での出力を予測するには、教師データが高々数百程度しか得られないため、データ効率が良いモデルが必要。
新規性
Bravais-Inspired Gradient Domain Machine Learning (BIGDML)を提案。これは周期系の材料に対し、正確でデータ効率がよく、計算コストが小さいモデルである。物理法則、対称性をモデルに組み込み、データ効率を向上させている。
手法
データ作成
NVTアンサンブルのMD計算でデータを収集
モデル
Symmetric Gradient-Domain Machine Learning (sGDML)をもとに作成。エネルギーが並進、回転に対し不変であること、エネルギーが保存することを制約としている。
ガウシアンプロセス(GP)をベースとしている。カーネルは普通のMaternカーネル。
GPに入力する特徴量は、クーロンマトリックスで作成。周期境界を考えるときに実質的な距離ではなく、並進させた場所での最小距離を採用する(合ってる?)。これにより長距離の相互作用を取り込める。これをカーネル関数で変換したのち、線形変換しForceを得る。線形変換のパラメータは学習される。この定式化により力場の積分が可能で、PESが構築される。
共分散行列を計算する際に、様々な対称変換(並進 × 回転・反転)に対し周辺化することで、対称性をモデルに組み込んでいる。

これらの効果を取り込むことで以下の図のように周期的に正しい描像が得られる。それぞれの要素を取り込んだときの精度の向上も顕著

結果
グラフェン
わずか10サンプルでこれまでのSOTAに匹敵する精度。GAP/SOAPでの学習と比較して10倍の精度。
表面データ
学習データが存在しないsaddle pointのエネルギーも正確に予測できた。

phonon
100個以下のデータセットであるにもかかわらず、DFTでの計算結果とほぼ一致。

課題
相変化などの対称性が変わる系への適用。
原子数が大きい系への対応(メモリ不足)
参考文献
[2022] A Universal Graph Deep Learning Interatomic Potential for the Periodic Table
概要
Materials projectの構造緩和データを大規模に学習したM3GNetの提案。MEGNetをベースに角度情報などの3体間の相互作用を取り込んだアーキテクチャ。3100万件の仮想結晶構造をスクリーニングし、180万件の安定に存在しうる構造を特定。
論文リンク
https://arxiv.org/abs/2202.02450
著者/所属機関
Chi Chen, Shyue Ping Ong
投稿日付
2022/2/5
背景
- 機械学習ポテンシャル(NNP)はユニバーサルに使えるものがなかった。
新規性
- 3体間相互作用を備えたGNNモデルでmaterials projectの巨大な構造緩和データを学習。
手法
-
1万8700件のenergy, 1600万件のforce、160万件のstressを学習
-
元素数は89
-
モデルのアーキテクチャは以下

-
これまではedgeは原子間の距離を基底関数で展開したものを用いていた。これはpropertyの予測には良いが、エネルギー曲面(PES)の予測には向かない。M3GNetではノードに原子の情報、エッジに原子間の結合情報、global state informationに温度、圧力などを与える。さらに差別化ポイントとして原子座標と格子定数を与える。
-
edgeの更新時にはその原子が属する全ての結合の情報(相手のノード情報と距離ベクトル)を考慮する。これにより3体相互作用を表現する。ベクトルを用いるため、距離、角度、2面角などの情報が含まれる。n体間の相互作用は層を重ねることで得られる。
-
角度情報は球面ベッセル関数と球面調和関数を用いて表現。

-
それぞれの特徴量は以下のように更新される。

この辺りはMEGNetと同じ? -
energy, force, stressをロスにして学習
-
1回目の構造緩和の最初及び中間の構造、2回目の構造緩和の最後のステップを学習データとして取得。最終のエネルギーが50 meV/atomを超えるもの、原子間距離が0.5Å以下のものは除外。分布が以下のようなデータセットを作成。

反発力を学習させるため、原子間距離が小さいものもデータセットに不可欠。
結果
-
energyだけで学習したものはforceの予測精度が悪かった。stressに関しても、energy+forceで学習したものと比較して、stressも追加して学習したものは半分程度のMAEになった。それぞれのMAEは0.035 eV/atom, 0.072 eV/Å, 0.41 GPaであった。

値が小さいところを学習するのはやはり難しそう。実際、弾性率の予測精度はかなり悪い。

-
M3GNetを用いて緩和した構造のエネルギーと基底状態のエネルギーのMAEは40 meV/atom程度。DFTで緩和した場合とほぼ同程度。
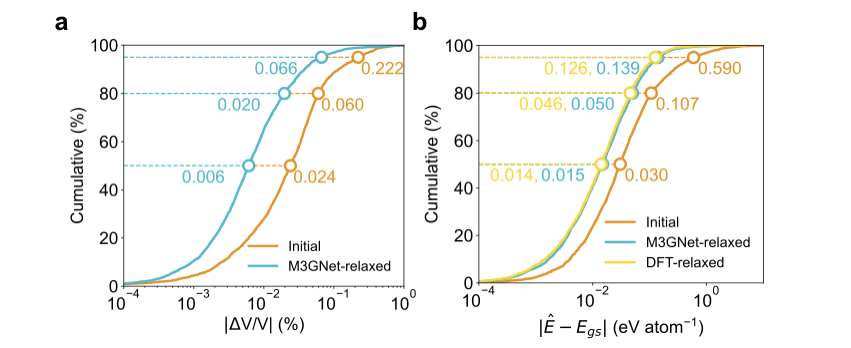
-
3100万件の仮想結晶構造をスクリーニングし、180万件の安定に存在しうる構造を特定。さらにtop1000件の構造に対しDFTでEhullを計算したところ、999件は1 meV/atomであった。酸化物に限った場合は1, 5, 10 meV/atomの閾値内に入った件数はそれぞれ579, 826, 935件であった。

-
Li3YCl6のアレニウスプロットがDFTとほぼ一致
-
MD-17ベンチマーク

Future work
-
データセットはラフな精度の構造緩和ではなく、1点計算を正確に行ったものが望ましい。materials projectでは今後、そのようなデータセットを作る検討をしている。
-
NNPで安定であると予測されたデータをDFTで緩和し、フィードバックするなどのactive learningの枠組みを取り入れる。これは特に未知領域の探索時に効率的。
参考文献
[2022] Artificial neural network approach for multiphase segmentation of battery electrode nano-CT images
概要
電池のX線CT画像のセグメンテーション。SegNetをベースにしたモデルで学習し90%程度の認識精度を達成。他のデータセットへの転移学習も有効。一方で、特に境界において作業者によるラベリング誤差が相当に大きく、精度向上を妨げていることを示し、これらの誤差を無くすことが重要と主張している。
論文リンク
論文:
https://www.nature.com/articles/s41524-022-00709-7
データ:
https://tomobank.readthedocs.io/
コード:
https://segmentpy.readthedocs.io/
著者/所属機関
@Article{su2022artificial,
title={Artificial neural network approach for multiphase segmentation of battery electrode nano-CT images},
author={Su, Zeliang and Decenci{`e}re, Etienne and Nguyen, Tuan-Tu and El-Amiry, Kaoutar and De Andrade, Vincent and Franco, Alejandro A and Demorti{`e}re, Arnaud},
journal={npj Computational Materials},
volume={8},
number={1},
pages={1--11},
year={2022},
publisher={Nature Publishing Group}
}
投稿日付
2022/2/9
背景
-
X線CT(XCT)画像は三次元構造情報を取得するための有用なツールであり、得られた情報をもとに電気化学シミュレーションを行うことで電極の特性を知ることができる。
-
電極のXCTデータでは単純な輝度の2値化やk-meansを用いたクラスタリングでセグメンテーションを行うことは難しく、別の手法が必要である。
-
他分野では深層学習によるセグメンテーションが盛んに研究されている。それらの例と異なり、電池材料では、結晶、凝集体、ポリマー、空隙が複雑な形状をしている。
新規性
- 電池のXCTデータに対し深層学習によるセグメンテーションを実施
手法
- モデルのアーキテクチャは以下。SegNetをベースにしたモデルになっている。XCTデータは量が多いので、パラメータ数を控えめにして推論速度を上げている。

- ランダムクロップとガウシアンノイズの付与でデータ拡張を行った。
結果
-
90%程度の精度を獲得。数十億のボクセルデータを数分で予測可能。
-
ラベルをつける人の認識バイアスにより、データのラベルにはサブセットごとに、ある程度の誤差があり、精度はある時点で飽和する。ある研究では、作業者の差に起因する誤差は少なくとも10%程度あることが報告されている。
以下は異なる作業者でのラベルの誤差。特に境界において大きな誤差が出ている。

この誤差は最も良いモデルの誤差と同程度のレベルであり、人間の認識誤差を取り除く必要があると主張している。ランダムに境界を変化させたデータを生成し、これらの誤差を定量化した。

- 他のデータセットに対して転移学習が有効であることも示した。
参考文献
-
NCMのセグメンテーション
Jiang, Z. et al. Machine-learning-revealed statistics of the particle-carbon/binder detachment in lithium-ion battery cathodes. Nat. Commun. 11, 2310 (2020). -
グラファイトのセグメンテーション(不確実度の値あり)
LaBonte, T., Martinez, C. & Roberts, S. A. We Know Where We Don’t Know: 3D
Bayesian CNNs for Uncertainty Quantification of Binary Segmentations for
Material Simulations. Preprint at https://arxiv.org/abs/1910.10793.
[2022] Pre-training via Denoising for Molecular Property Prediction
概要
安定構造にガウスノイズを加えた構造のデノイジングを学習することで、分子の物性予測に有効な事前学習が可能であることを示した。これは局所解周りの力場の学習に相当する。QM9の全てのタスクにおいて事前学習により精度が向上し、12個中10個のタスクでSOTA更新。
論文リンク
https://arxiv.org/abs/2206.00133
著者/所属機関
Oxford Univ., DeepMind
Zaidi, Sheheryar and Schaarschmidt, Michael and Martens, James and Kim, Hyunjik and Teh, Yee Whye and Sanchez-Gonzalez, Alvaro and Battaglia, Peter and Pascanu, Razvan and Godwin, Jonathan
投稿日付
2022/5/31
背景
-
NNによる分子の物性予測は、データ数が少なく適用が限られていた。
-
QM9データセットのSOTAモデルは事前学習を行ったモデルではなく、最適な事前学習が行われていない。
-
分子の事前学習の多くは2次元のグラフを対象にしていた。GraphMVPは3次元構造を取り入れているが、下流タスクは2次元の情報に関するもの。
-
著者のグループでは、過去にNoisyNodesを提案している。デノイジングを補助タスクとして解くことで性能を向上させることができ、分子の特性予測に有効であることを示している。
新規性
-
平衡状態の構造を用いた事前学習手法を提案。構造にガウスノイズを加え、ノイズ除去の学習を行う。これにより力場の一部を学習でき、分子表現に有用な特徴を獲得可能である。
-
事前学習に関してデータ数、モデルサイズ、アーキテクチャの影響を調査
手法
事前学習
分子内の各原子位置に対しガウスノイズを加える。これをGNNに通した時の出力値とノイズの差分を最小化する(ノイズを学習する)。

分子の物性予測にはenergyやforceが大きく関与することから、力場の学習は良い事前学習になる。ボルツマン分布の近似として混合ガウス分布を導入する。このスコアマッチングは力場を学習することに相当する。また、過去の報告よりこれはデノイジングと同等であることから、上記の関数を学習することで力場を学習できる。ガウシアンの幅は実際の力場の局所解周りのシャープさに相当するが、決め方についての言及は無い。
デノイジングを目的関数とすることで、GNNのover-smoothingが抑制できる(出力の多様性が求められるため)。
モデル
-
メインのモデルとしてGNSと、それを改良したGNS-TATを用いた。GNSはエンコーダとデコーダを備えている。
-
エンコーダではノード特徴量(原子のembedding)とエッジ特徴量(原子のベクトル)を更新していく。プロセッサーで、初期のグラフと同等の接続を持つグラフを生成。ノード、エッジ特徴はいくつかの特徴を結合しMLPで変換することにより更新される。
-
デコーダでは更新されたノード特徴量をそれぞれ別個にMLPで更新(ノード間の相互作用はない)。グラフレベルの特徴量が必要な場合は最後にaggregateする。

データセット
事前学習用のデータセットはPCQM4Mv2あるいはOC20を使用。下流タスクのデータセットはQM9, OC20, DES15Kを使用。データ分布が最も異なるのはPCQM4Mv2→OC20のタスク。
結果
OM9
QM9データセットで12個のタスク中10個でSOTAを記録。

事前学習を行うことで全ての物性に対し精度が向上している。<R^2>タスクで性能が悪いのは、最適なノイズのスケールが他のタスクと異なることに起因していると考えられる。
OC20
事前学習のデータセットにPCQM4Mv2を用いた時は性能の向上は見られないが、OC20を事前学習のデータセットとして用いると、収束までのステップ数が小さくなった。ただし最終的な性能はあまり変わらない。Noisy Nodeを補助タスクとして解くことと、デノイジングの事前学習はこのケースにおいては同等であるため。

DES15K
事前学習を行うことで精度が向上。

これにより、計算コストの低い手法でのデータを用いた事前学習は、より高いコストのデータに対する転移もうまくいくことが示された。
事前学習条件の検討
-
モデルをTorchMD-NETに変更しても事前学習は有効であることが示された。ただし、不安定である傾向が見られ、検証中とのこと。
-
事前学習、fine-tuning時の学習データ数を変えると、いずれもデータ数が増えるほど性能が改善した。また、モデルのパラメータ数を増加させても性能が向上した。

- デコーダのみをfine-tuningすると、スクラッチより性能が大幅に高いことが観測された。これにより、事前学習で良い特徴が得られていることが示された。
課題
- 分子の物性予測に用いられるアーキテクチャはNLPと異なり標準化されておらず、事前学習した重みを有効に使えない可能性がある。
- データ分布のどの特性が下流タスクと一致すべきかが定かではない。
- モデルのアーキテクチャによっては事前学習が安定しない。
参考文献
[2022] Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements
概要
45元素に適用可能な汎用なNNPを構築。従来のデータセットには含まれない不安定構造やイレギュラーな構造も含めて大規模に学習し、45元素内であればドメイン外の構造にも高い汎化性能を示した。Li正極材料、MOF、合金、触媒などでの実応用でも高い精度を示すことが確認された。
論文リンク
https://www.nature.com/articles/s41467-022-30687-9
著者/所属機関
@Article{takamoto2022towards,
title={Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements},
author={Takamoto, So and Shinagawa, Chikashi and Motoki, Daisuke and Nakago, Kosuke and Li, Wenwen and Kurata, Iori and Watanabe, Taku and Yayama, Yoshihiro and Iriguchi, Hiroki and Asano, Yusuke and others},
journal={Nature Communications},
volume={13},
number={1},
pages={1--11},
year={2022},
publisher={Nature Publishing Group}
}
投稿日付
2022/5/30
背景
-
DFTの高速化技術としてニューラルネットワークポテンシャル(NNP)が注目されている。QM9、Materials Projectなどのデータセットがあるが、既存材料に限っているため、未知材料に対する予測が困難であった。また、これらは局所安定構造のデータセットであるため、化学反応や拡散、相転移などのポテンシャル予測には適さない。そのため、材料探索という観点ではこれまでのNNPデータセットでは大きな問題があった。
-
ANI-1やANI-2xは安定構造から変位した構造も含んだデータセットであるが、分子のみのデータセットであり、結晶構造や表面のデータを含まない。
-
Open Catalyst(OC20)は2億超の巨大なデータセットであるが、触媒反応の表面データしか含まれない。さらにこれらの触媒材料は既存材料のみを対象としている。
-
つまりこれまでのデータセットは一部のドメインのみを含んだものであり、様々なドメインをカバーしたデータセットではなく、未知の構造の予測精度が低かった。
-
NNは多くの分野でデータセットやモデルサイズと精度の間に冪乗則が確認されている点と、NNは多様なデータに対する表現力を十分に有していることから、膨大なデータセットと適切なアーキテクチャを用いることで従来のNNPの問題点を解決できることが示唆される。
新規性
-
既存構造のみならず、不安定な構造を積極的に収集し、汎化性能を高めた。さらに通常観測されないような元素ペアの構造を様々な密度で生成しデータセットに加えた。
-
45元素の任意の元素ペアに対しシミュレーション可能なポテンシャル(PFP)を作成
手法
データセット
- 分子、結晶、クラスター、表面、歪んだ結晶など、様々な構造を含んだデータセットを構築。

- それぞれのデータセットの統計量は以下。

-
DFTでEnergyとForceを計算。結晶データはatomic chargeも算出。
-
分子データはH, C, N, O, P, S, F, Cl, Brからなる構造。不安定な分子やラジカルも含む。1つの構造から、構造緩和、NMS(Normal Mode Sampling), 高温MDなどで複数の構造を取得し、データセットに加えた。H-Krまでのほぼ全ての組み合わせに対し2体間ポテンシャルを計算した。
-
結晶データセットは45元素を対象とした。例えばSiについて、他の不安定な構造(Cubic_Pm3m, FCC_Fm3m, NaCl-type_F43m)などもデータを取得。バルクとクラスターに関してはセルの体積や形状を変化させた構造や、ランダムに原子位置をスワップした構造も生成。結晶ではNMSは用いなかった。
表面データセットは分子をランダムに配置したデータと、それをPFPで緩和した構造を生成。
歪んだ構造のデータセットではランダムに選択•配置した原子からなる構造を高温でMD計算しデータを生成。H-Biまでのほぼ全ての組み合わせに対し2体間ポテンシャルを計算した。 -
データセットの一部はHME21データセットとして公開されている。
データセット生成方法
molecule
- GBD-11、PubChemから分子を取得。さらに原子を修飾(置換?)した構造を生成。これらをベースの分子とする。2体間ポテンシャルの構築のため全てのペアの2原子分子も含まれる。構造緩和、NMF、MDでこれらの分子から複数の構造を生成。2分子からなる構造もMDで取得。
bulk
45の元素から構成される1元系、2元系の構造をベースの結晶として生成。
1元系に関してはpc, fcc, bcc, hcp, ダイヤモンド構造、一次元鎖、2次元グラフェン構造を生成。
2元系に関しては全ての元素ペアに対し閃亜鉛鉱、岩塩、塩化セシウム、ウルツ鉱の構造を生成。さらにMaterials Projectから、16原子以下で遷移金属を含む構造を取得。
これらのベース構造を、DFTでセル形状と原子の分率座標を固定したままま体積のみ緩和。これらのベース構造に対し4つの変位を与えた構造を生成。
-
セルの形状と原子の分率座標を固定したままセルの体積を変化。計20構造。セルの長さを1.0-1.1倍した構造を10個、セルの長さを1.0-1/1.1倍した構造を10個生成
-
セルを固定したまま原子位置を変化。0-0.1の均一分布からサンプリングした値にサイト間の平均距離をかけたものでそれぞれのサイトの変位距離をサンプリング。方向はランダムにする。計10構造
-
剪断変形。セルの体積と原子の分率座標を固定したまま剪断変形する。α,β,γから1つ選択し、-5から+5°の間で等間隔な値21個を取得し変化させる。(0°は意味ないので20個)。α,β,γ全てに対し適用。計60構造。(ここから同一の構造は削除するため少し減る)
-
引張変形。セルの体積、角度、原子の分率座標を固定したまま引張変形する。a,b,cから1つ選択し0.9-1.1から等間隔の値21個でスケールする。他の2つの軸も体積を固定するため変化させる。計60構造。(ここから同一の構造は削除するため少し減る)
cluster
単一元素からなる13原子のクラスターを作成。原子間距離は共有結合半径を使用。真空層を10Å入れた構造を生成。
disordered
高温MDで生成。MDを実行するためのポテンシャルはdisorderedを含まないデータセットで作成したPFPを使用。
- cubicのセル(8.8(主) or 8.4 or 8.0Å)を作成
- 2×2×2のfcc構造として、セル内に原子を配置。これらの原子は以下のいずれか
(a): ランダムに抽出。元素の最大数は20に設定。
(b): 2 or 4 or 8元素を繰り返し選択する。選択された元素は4つの原子をセルに配置する。32原子になるまで2,4,8元素の選択を繰り返す。
(c): (b)の手法で生成し、最大24原子まで削除する。(疎な構造を作る) - 高温MDを実施。原子の質量を5(atomic mass unit)に統一。6000Kを初期温度とし、1ps分NPTアンサンブルで10000KでのMDを実施。2000K, 1ps → 500K, 1psでNPTのMDを実施。
- 1つの軸方向に真空層を入れ、300, 1ps分NPTでMDを実施
- それぞれの最終構造(全4構造)を取得。ただしDFT計算でForceが20 eV/Åを超えるものは除いた。
slab
(111), (110), (101), (011), (100), (010), (001)表面を作成。真空層は10Åに設定。bulkと同一の構造変形を実施。(生成構造数は半分にした。)
adsorption
スラブと分子をランダムに取得。2パターンの構造を生成。
(a)構造緩和:
分子はランダムに回転し、スラブのランダムな表面位置に配置する。これをPFP(disorderedを除いたデータで学習したもの)で緩和する。この時スラブは全て固定。変形させていないスラブに対し適用。
(b)ランダム配置:
(a)と同様に分子を配置し、スラブと分子の最小規格化距離が0.8-1.3のランダムな値になるように垂直に移動させる。変形していないスラブと、セル膨張•収縮させたスラブに対し適用。
DFT計算条件
分子
ωB97X-D交換相関汎関数、6-31G(d)基底関数を使用。singletとdoubletのみ考慮。
結晶
- PBE交換相関汎関数、energy cutoff 520 eV、スピンあり、GGA+Uで計算。
- 金属や半導体、絶縁体を同一の条件で計算するため、gaussian smearing(σ=0.05 eV) で計算。また、金属のみの場合でもGGA+Uで計算。
- 強磁性、反強磁性の考慮のため平行、反平行の初期配置から計算し、エネルギーが低いものを採用。ただし系によっては平行初期値のみ。
- Bader chargeも計算。
モデルの学習
- 作成したデータセットに加え、OC20データも同時に学習させた。計算条件を特徴量として加え学習。推論時には計算条件の指定が可能。
- energy, forces, atomic chargeを学習。今回計算したデータのエネルギーとOC20のエネルギーの基準は合わせているか不明。
モデルアーキテクチャ
- TeaNetをベースに作成。これはtensorをequivaliantな層で処理したGNNモデル。
- Morse potentialとの差分を学習することでPESを物理的に妥当になるように制約している(データセットにない近距離の原子の反発力を記述できる)。全てのペアに対しパラメータを学習している。
- 電荷を出力するが、この電荷を用いて算出されるクーロン力を取り入れることはしていない。
- E(3)-invarianceなアーキテクチャ。(エネルギーに対してということだと思う。)
- GNNの層数は5で、計算コスト削減のため3,3,4,6,6Åの異なるカットオフを採用している。
- 高次元の微分値を持つように設計。Newton法などの最適化時や弾性率の計算時などに必要になる。
- 他の代表的なモデルとの比較

NequIPは今はE(3) equivariantなはず。
結果
モデルパフォーマンス
- テストデータに対する予測結果。disorderedな構造に対しても精度が高い。

- フェノールとフェーノールにLi or Naを配位させたものをcrystalモードとmoleculeモードで予測。含有元素はcrystalモードはドメイン内のデータだが、moleculeモードではドメイン外のデータになる。moleculeモードにおいてもcrystalと同等の精度が出ている。データセットを混合することで、あるDFT計算条件のデータの外挿領域でも、他の計算条件のデータセットの内挿領域であれば予測が可能になる。これにより、今後計算条件が異なる多様なデータセットが出てきても、それらを同時に学習することで相補的にデータを拡張することができる。
応用例
- LiFeSO4FのLi拡散の活性化エネルギーを正確に予測。LiFeSO4Fのデータや反応遷移状態のデータは、学習データに入っていないにもかかわらず高い精度で予測できた。
- MOFデータに関して、構造緩和や水との結合エネルギー評価が正確であった。ちなみにMOFデータは学習データに入っていない。
- CU-Au系合金のorder-disorder transitionを正確に予測。
- Fischer-Tropsch触媒の探索を実施。Co(0001)表面における合成ガスのメタネーション反応の活性化エネルギーがDFTでの値とほぼ一致(MAE=0.097 eV)。また、FT反応で重要なCOの乖離反応を促進する添加元素を探索。Vが反応を促進することが観測され、実験と一致した。
参考文献
[2021] Pre-training Molecular Graph Representation with 3D Geometry
概要
分子の三次元構造情報を取り込んだ表現学習手法GraphMVPの提案。contrastive-SSLとgenerative-SSLを組み合わせることで、従来の2次元のトポロジーのみの表現学習ではなく、3次元の情報(エネルギーの情報が含まれる)を取り入れている。8個の下流タスクにおいて他の表現学習モデルを用いた場合と比較して高い精度が得られた。

論文リンク
https://openreview.net/forum?id=xQUe1pOKPam
著者/所属機関
Shengchao Liu1,2, Hanchen Wang3, Weiyang Liu3,4, Joan Lasenby3, Hongyu Guo5, Jian Tang1,6,7
1Mila 2Université de Montréal 3University of Cambridge 4MPI for Intelligent Systems, Tübingen 5National Research Council Canada 6HEC Montréal 7CIFAR AI Chair
投稿日付
2021/9/29
背景
- 分子の特性を予測するためには3次元構造が重要だが、現実世界で得られる3次元構造のデータ数は限られている。そのため3次元データを用いた下流タスクは限られている。
新規性
- 2次元構造と3次元を結びつけるGraphMVPを提案。これにより下流タスクでのファインチューニング時には3次元構造無しに高い性能が得られる。3次元構造の情報を2次元構造のグラフエンコーダーに入れ込むように学習を行う。
- contrastive-SSLとgenerative-SSLを組み合わせた表現学習の手法を提案
手法
-
分子間レベルの学習:対照学習を使う。2Dと3Dのグラフが同じ分子から得られたものであれば正例、そうでなければ負例にラベリングする。
-
分子内レベルの学習:2D/3Dの表現から3D/2Dを作る。これにより3次元構造の情報を2次元グラフに取り込む。生成モデルはGAN, VAE, flow-baseなどの候補があるが本論文ではVAEを用いた。生成の精度を定量化するためにVRRという指標を導入。これは2次元グラフのエンコーダーで得られた潜在表現を直接三次元グラフの潜在空間に投影し誤差を評価するということっぽい。
-
3Dはエネルギー、2Dはトポロジーに焦点を当てた表現であり、これらを結びつけたいという**
-
2次元グラフはノード特徴量とエッジ特徴量からなり、3次元グラフはノード特徴量と座標からなる。それぞれGIN, SchNetを使用
結果
-
8個の下流タスクにおいて他の表現学習モデルを用いた場合と比較して高い精度が得られた。
-
conformer数を増やすと精度が上がったが、上昇率は低かった。これは多くのケースで平衡状態のconformerはtop5程度で十分であることを示唆している。
-
今後はよりパワフルな表現方法を試したり、より大規模な分子やタンパク質に適用予定。
参考文献
[2022] MatSciBERT: A materials domain language model for text mining and information extraction
概要
材料に特化したBERT(MatSciBERT)の論文。データ数が少量であることに対応するため、生命医学とコンピュータサイエンス分野で事前学習したSciBERTの重みで初期化し、材料の論文で事前学習を実施。様々な下流タスクでSciBERTやBERT、他のベースラインモデルよりも高精度。利用シーンは論文分類・クラスタリング、ナレッジグラフ構築、合成経路予測など。
論文リンク
https://www.nature.com/articles/s41524-022-00784-w
コード:https://github.com/M3RG-IITD/MatSciBERT
モデル:https://huggingface.co/m3rg-iitd/matscibert
著者/所属機関
@Article{gupta2022matscibert,
title={MatSciBERT: A materials domain language model for text mining and information extraction},
author={Gupta, Tanishq and Zaki, Mohd and Krishnan, NM and others},
journal={npj Computational Materials},
volume={8},
number={1},
pages={1--11},
year={2022},
publisher={Nature Publishing Group}
}
投稿日付
2022/4/28
背景
-
科学のテキストから情報を抽出するのは、複数のパラグラフにまたがって参照されていたり、図のキャプションであったりなどの理由で難しい。
-
科学分野ではChemDataExtractorで論文などのテキストから情報を抽出する例がある。他にはOSCAR4やArtifical Chemistなど。
-
BERTの特定ドメインへの適用例はBioBERTやSciBERT(生命医学とコンピュータサイエンス分野のモデル)、clinical-BERT、mBERT、PatentBERT、FinBERTなどがある。しかし、材料分野に特化したBERTモデルはなかった。
新規性
- 材料分野に特化したBERTモデルを作成。自動で情報を抽出、ナレッジグラフの作成、あるいは他の下流タスクへの適用に有用なモデルを作ることを目的とする。
手法
-
材料に適したコーパスとして、無機ガラス、金属ガラス、合金、セメント、コンクリートのコーパスを作成。
-
Elsevier Science Direct Databaseの100万件のうち15万件の論文を取得。本文を全てコーパスに入れた。500件の論文に対し人力でアノテーションし、SciBERTをファインチューニングした。学習済みのSciBERTで残りの論文のアノテーションを行った。
-
コーパス内の単語は285M個であり、これはSciBERT(3.17B)やBERT(3.3B)の9%程度であった。SciBERTを初期にして事前学習をするので、実質的には3.45Bの単語数。
-
大規模の事前学習モデルをさらに特定分野で事前学習すると性能が高くなることが知られているため、語彙の重複が多かったSciBERTの重みで初期化した。事前学習はRoBERTaと同様の方法で行った。(単語全体のマスク、NSP lossの除去、文全体のみでの学習、大きなバッチサイズ)。GPU2台で15日間の学習を行った。
-
Named Entity Recognition (NER), Relation Classification, Paper Abstract Classificationの3つの下流タスクを実施。それぞれ、単語の分類、単語間の関係の分類、アブストの分類のようなもの。
結果
- MatSciBERTとSciBERTとBERTでNERの下流タスクの性能を評価したところ、いずれのデータセットにおいてもMatSciBERTが最も高い性能を示した。

材料特有のentityに対しては全てSciBERTを上回っており、材料コーパスでの学習で、材料に対する知識が得られていることが分かる。

-
Materials Synthesis Procedures datasetのRelation ClassificationタスクやPaper Abstract ClassificationタスクでもSciBERT, BERTおよび他のベースラインモデル(MaxPool, MaxAtt)より高い性能を示した。
-
MatSciBERTは以下のような利用シーンが想定される。
-
論文の分類:従来はTFIDFやWord2Vecが使われていたが、単語単位での認識のため性能が低い。MatSciBERTでは文脈を理解できるので性能が高い。
-
類似トピックのグループ化:これもTFIDFやWord2Vecをもとにしているので文脈を考慮できないがMatSciBERTでは可能。
-
画像からの情報取得:キャプションには多くの情報が含まれるが、entityを正しく理解したモデルでないと情報を取得できない。MatSciBERTはNERタスクで好成績なので、情報が取得可能。
-
材料分野でのナレッジグラフ構築:2-hopのentity間の相関を見るなどで、ナレッジグラフが構築できる。

- その他:関係性分類や質問応答などに適用可能。関係性分類では最適な合成経路予測に応用可能
- 材料データでは生命医学と比較して少量のデータセットしか取得できないので、より多くのデータセットの整備が求められる。
参考文献
-
NLPで自動作成した電池データベース
Huang, S. & Cole, J. M. A database of battery materials auto-generated using ChemDataExtractor. Sci. Data 7, 260 (2020). -
NLPでの合成ルート予測
Epps, R. W. et al. Artificial chemist: an autonomous quantum dot synthesis bot.
Adv. Mater. 32, 2001626 (2020).
[2022] MetaRF: Differentiable Random Forest for Reaction Yield Prediction with a Few Trails
概要
反応の収率予測において、ランダムフォレストにNNを合わせた微分可能なモデル(MetaRF)を活用。メタ学習でNNの初期重みを自動で決定することで、few-shot学習において通常のRFよりも高い性能を示した。ablation studtyにより、RFはfew-shot学習時の過学習抑制に重要であることが判明。
論文リンク
https://arxiv.org/abs/2208.10083
著者/所属機関
Chen, Kexin and Chen, Guangyong and Li, Junyou and Huang, Yuansheng and Heng, Pheng-Ann
投稿日付
2022/8/22
背景
合成経路予測(CASP)はデータセットが大量に整備されつつあり、多くの成果が得られている。しかし、収率の予測なしには、これらは実際に採用にまで至らない。
一般に最適経路予測はOOD問題である。これまでランダムフォレストでOODデータに対し高い性能を示すモデルが報告されてきたが、収率予測ではOODデータの予測精度は悪かった。
新規性
-
少数データ(5個程度)を加えることを許可し、few-shotでの収率予測を実施。
-
ランダムフォレストにNNを合わせた微分可能なモデル(MetaRF)を実装。メタ学習でNNの初期重みを自動で決定する
手法
特徴量
DFT特徴量(分子、原子、振動に関する特徴量らしい。DFTで計算する?)を用いて各基質をエンコードし、結合することで反応の特徴量を得た。
メタ学習
- メタ学習のフレームワークとしてMAMLを用いた。これはfew-shot学習時に高い性能が得られるようにモデルの初期重みを最適化する手法。各タスクにおける、数回重みを更新した後の目的関数を足し合わせ、それが最小になるように初期重みを最適化する。つまり、さまざまなタスクに適用可能な初期重みを獲得することを目的とする。
本研究では、メタ学習とランダムフォレストを組みあわせるため、ランダムフォレストで得られた出力群(各決定木の出力)を小さなニューラルネットワークに流し、出力を得る。この重みの初期値をメタ学習で最適化する。一回更新後の重みを用いて目的関数の和を評価した。
メタ学習で用いるタスクでは、添加剤をサンプリングしその添加剤が含まれる反応の中からいくつかサンプリングしたターゲットの収率予測を行う。

サンプリング
- few-shot学習では、few-shot学習時のサンプルの選び方は精度に大きく影響する。本研究ではKennard-Stone(KS)アルゴリズムを用いてサンプリングを行なった。特徴量空間をTSNEで次元削減し、KSアルゴリズムによるサンプリングを実施した。
結果
- MetaRFはベースラインモデル(通常のランダムフォレスト)と比較して、高い精度が得られた。

MetaRFは少数のデータでも高い性能が得られている。
また、それぞれのモデルを用いて、最も収率が高い反応のtop10を予測させ、それらの平均と標準偏差を計算すると、MetaRFはベースラインモデルよりも高い性能を示した。

- 以下の3つのablation studyを実施
- サンプリングをランダムにする2.
- ランダムフォレスト部分を無くす(MAMLでNNだけのモデルの重みの初期値を学習?)
- メタ学習ではなく転移学習にする

いずれも精度が低下。特にランダムフォレストはfew-shot学習時の過学習抑制に大きく寄与していることが判明。転移学習もベースラインよりは精度が上がるが、メタ学習の方がより高い精度が得られた。
- 特徴量の重要度は以下。

参考文献
[2021] SE(3)-Equivariant Graph Neural Networks for Data-Efficient and Accurate Interatomic Potentials
概要
エネルギー、フォース予測において、SE(3)同変な層により構成されたGNNモデル。実はE(3)同変も対応している。ベクトルやより高次なテンソルの入力が可能になり表現力アップ。様々なベンチマークでSOTA更新

論文リンク
https://arxiv.org/abs/2101.03164
著者/所属機関
Simon Batzner, Albert Musaelian, Lixin Sun, Mario Geiger, Jonathan P. Mailoa, Mordechai Kornbluth, Nicola Molinari, Tess E. Smidt, Boris Kozinsky
投稿日付
2021/2/8
背景
従来の機械学習ポテンシャルはデータ効率が悪く多くのデータが必要であった。一方、カーネルベースの機械学習法は精度が悪かった。
新規性
SE(3)同変な層でネットワークを構成することによりデータ効率が高く、高精度な機械学習ポテンシャルモデルを提供。
手法
-
相対位置を距離ではなくベクトルで表すことで回転に対し同変な特徴を得る。これにより角度の特徴を取り込むことが可能。ちなみにDimeNetは角度の情報が入っているが、スカラーとして入力しているため、同変な量になっていない。
-
ネットワークは同変な層から構成される。動径関数、球面調和関数、クレプシュゴルダン係数によりテンソル積を処理する。
-
Forceはenergyの微分で計算する。同変なネットワークなので直接予測することも可能だが、MDでエネルギー保存則が保証されないので、本研究では微分で算出。
-
原子はスカラー、ベクトル、テンソルの特徴量で記述される。これらは規約表現の和で表される。
結果
-
MD17でSoTA更新

-
Li-P-OのMDをよく再現
-
データ効率が高い

参考文献
[2022] Periodic Graph Transformers for Crystal Material Property Prediction
概要
結晶構造の周期的な情報を取り入れたMatformerを提案。各ノードに対し、隣接セルの同一原子とのエッジを導入し、明示的にセルの形、大きさを取り入れる。エッジごとのattentionを取り入れたtransformer型のアーキテクチャを採用。他の無機系モデルに比べ高精度を実現。
論文リンク
https://arxiv.org/abs/2209.11807
著者/所属機関
Yan, Keqiang and Liu, Yi and Lin, Yuchao and Ji, Shuiwang
投稿日付
2022/9/23
背景
結晶構造ではE(3)-invariantであるのに加え、周期境界に対してもinvariantでないとならない。セルを繰り返したときの不変性は考慮されているものが多いが、セルの平行移動に対しての不変性は考慮されていないものが多い。周期的なパターンを明示的に取り込むことで結晶構造の適切な表現ができると考えられる
新規性
periodic invarianceと周期的パターンのエンコーディングをtransformerに明示的に取り入れたMatformerを提案。
手法
従来のグラフ作成方法では以下のように隣接原子との間の情報が得られるだけ。セルの形や大きさがどのようであるかの情報が欠落している。

Matformerでは以下のように、隣のセル内にある同じ原子に対しエッジを作る。これはセルの形と大きさの情報を明示的に導入することに対応する。

グラフを作るときの注意点として、カットオフで隣接原子を取った後、隣接原子の数を制限することは避けないといけない。そうしてしまうとperiodic invarianceが破綻してしまう(同じ距離に違う原子があり、どちらかしか選択されなかった場合など)。CGCNNなどではこのような操作をしているため、好ましくない。
ネットワークの全体像は以下

モデルの表現力向上と異なる次元(エッジ数?)のノードを区別するため、attentionのsoftmaxは無くしたとのこと。先行研究のGraphormerではノードごとのattentionだが、Matformerではエッジごとのattentionにした。
結果
ベンチマーク用のモデルでMPデータを再学習した(論文に報告の値はそれぞれデータセットが異なるため)結果が以下

全てのタスクにおいて最も高い性能を示した。
また、ALIGNNと比較して、高精度かつ高速に計算できる。

ablation sttudyにおいて、periodic invarianceとperiodic encodingがゆうこうであることが確認された。

角度情報を明示的に取り込んだバージョンも作成したが精度が向上しなかった。これはperiodic encodingで結晶構造を十分に表現できていることを示唆している。
参考文献
[2021] Direct Prediction of Phonon Density of States With Euclidean Neural Networks
概要
E(3)同変なGNNモデルを用いてphonon-DOSを予測する論文。E(3)同変性によりデータ効率がよく、1000件程度のスモールデータでも精度の高いモデルが構築できた。ノード特徴を質量を含むOne-hotベクトルで表現し、合金系にも適用可能にした。(Multi-hotで質量に係数をかけるだけでいい)

論文リンク
https://onlinelibrary.wiley.com/doi/10.1002/advs.202004214
著者/所属機関
Zhantao Chen, Nina Andrejevic, Tess Smidt, Zhiwei Ding, Qian Xu, Yen-Ting Chi, Quynh T. Nguyen, Ahmet Alatas, Jing Kong, and Mingda Li
投稿日付
2021/3/16
背景
-
限られた情報から、複雑で無限にある連続値を予測するのは難しい。決勝の場合はさらに対称性の制限がかけられる。
-
phononデータ(特に合金系)のデータは収集コストが高い。
新規性
-
phonon予測にE(3)同変のネットワークを用いた
-
ノードは原子質量を掛けたone-hotベクトル。合金系は原子質量に係数を掛けたmulti-hotベクトルで表すことが可能で、追加の学習を必要としない。
手法
-
Tensor-field-networkと同様のアーキテクチャ。追加でL1Netのゲート機構が入っている。動径関数、球面調和関数、クレプシュゴルダン係数でテンソルを取り扱う。
-
合金系のノード特徴量

結果
- testデータはvalデータと誤差が同程度で汎化性能が高い。

-
精度が低いものでも大まかな特徴は捉えている。
-
DFTが教師データのため、実験とはあまり合わないが、やはり大まかな特徴は捉えている。
-
単純な3次元CNNモデルで学習させるとかなり精度が低かった。E(3)同変性という結晶に必要な制約を入れることは精度向上のために必要である。
参考文献
-
phononデータベース
G. Petretto, S. Dwaraknath, H. P. Miranda, D. Winston, M. Gi- antomassi, M. J. Van Setten, X. Gonze, K. A. Persson, G. Hautier, G.-M. Rignanese, Sci. Data 2018, 5, 180065. -
L1Net(ゲートつきグラフ畳み込み)
B. K. Miller, M. Geiger, T. E. Smidt, F. Noé, arXiv e-prints, arXiv:2008.08461 2020.
[2022] Upper-Bound Energy Minimization to Search for Stable Functional Materials with Graph Neural Networks
概要
緩和前の構造を用いて材料の安定性を評価する手法。scale-invariantなGNNモデルを用いて、緩和前構造を入力とし、体積のみ緩和した構造のエネルギーを予測。この値が競合相より安定であれば、完全緩和した構造も安定である。1430万件の仮想構造に適用した結果、1707件が安定と予測され、実際にDFT計算でも1700件(>99%)が安定であることが確認された。
論文リンク
https://chemrxiv.org/engage/chemrxiv/article-details/632807ae2984c941066439ea
著者/所属機関
Jeffrey N. Law, Shubham Pandey, Prashun Gorai, Peter C. St. John
投稿日付
2022/9/19
背景
DFTによる構造緩和を行わずに、体積のスケールに対しinvariantなGNNモデルを使って材料の安定性を評価する研究がある。
新規性
以下の方法で、先行研究をよりロバストなアプローチにする。
- セルの体積のみを変化させてDFTで構造緩和したデータセットを作成。これは完全に緩和した構造のエネルギーの上限になる。
- 緩和後のエネルギー予測モデルはscale-invariantなGNNを用いる。
手法
GNNアーキテクチャ
- 元素のone-hot表現をノード特徴量、12個の隣接原子に対する距離をエッジ特徴量とする。
- それぞれの構造について、原子間の最小距離が1Åになるようにスケールさせる。これによりモデルは構造の体積に対しscale-invariantになる。
データセット
- ICSDの構造を元素種によらない組成比に分け、空間群とソートされたワイコフラベルを比較し、ユニークな構造セットを作成。これをプロトタイプとした(4000組成比、>13000構造)。
- プロトタイプを元素置換し、仮想構造を作成。価数が異なる置換も、置換後に電気的中性になる組み合わせがあれば許容する。
- 学習用データはこれらを構造緩和して作成。構造緩和の体積初期値はpymatgenのDLSを用いて予測した。異なる初期構造から緩和しても同一の構造になる場合があり、その場合はそれらの初期構造を除外。
- 緩和後のエネルギーが、簡単な線形回帰モデルでの予測値と大きく異なる場合はデータセットから除外。
- matminerで作成した構造のfingerprintが同一と判断されたものは除外
結果
- まず、検証として、緩和前の構造を入力とし、完全(セル形状、原子位置含め)に緩和した構造のエネルギーを予測した。ICSD構造に対しては予測がうまくいくが、仮想構造に対しては、MAEは0.13 eV/atomと大きかった。これは緩和前後で構造が大きく変わってしまい、初期構造の情報からでは予測がうまくいかないためであると考えられる。緩和前後でのfingerprint距離(構造がvolume以外変わっていないなら0になる)を測ると86%以上が0.1を超える値であったため、確かに緩和前後で構造が大きく変化している(b)。これはOQMDデータセット(a)とは対照的であり、粗悪な初期構造では、完全に緩和した構造のエネルギー予測は困難であることを示している。

- そのため、セルの体積のみを緩和させたデータセットを作成。過去の研究から、scale-invariantなGNNモデルは最適な体積とエネルギーを、セルの形と原子の分率座標から正確に予測できることが示唆されている。このエネルギーは完全に緩和した構造の上限値となるので、有用な参照値として使用することができる。もしこの値で既に安定であれば、完全に緩和した構造も安定であることが保証されている。

- 1430万件の仮想構造に対して評価したところ、2003件の構造が安定であると評価された。このとき、モデルの予測誤差を考慮して分解エネルギーが0.1 eV/atom以上のものを抽出した。これらの構造に対し、DFTで構造緩和(完全に緩和と体積のみ緩和)を実施。1707件が収束し、体積のみ緩和した構造はGNNでの予測値と良い一致を示した(a)。ほぼ全ての構造は完全に緩和した場合のDFT計算値よりもGNN予測値が大きくなっており、上限値として使えるという仮説を支持した(b)。1707件中1700件は実際にDFTのエネルギー値ベースで安定であると確かめられ、99%という高い予測精度を実現できた。

-
固体電解質の探索に活用。安定性に加え、エネルギーから電位窓を計算。さらに伝導度を決める特徴として伝導種が占める体積を考慮して候補を抽出。Li2HfN2は金属Liに対しても安定であると予測された(酸化側は1.2Vと弱いが)。
-
今回はプロトタイプを置換することで仮想構造を生成したが、より柔軟な生成モデルを用いて仮想構造を生成することで、より多くの安定な構造を発見できる可能性がある(プロトタイプから外れると学習データ分布から離れて精度が落ちる気はするが)。強化学習を用いてサーチスペースを拡張していく予定とのこと。
参考文献
[2021] GAETS: A Graph Autoencoder Time Series Approach Towards Battery Parameter Estimation
概要
グラフを用いることで、電池の測定データ(電流、電圧など)間の依存関係を取り込みながらそれらの時系列データを予測するGAETSの提案。さらにオートエンコーダーのロスを加える正則化により、既存手法より精度向上。
論文リンク
https://arxiv.org/abs/2111.09314
著者/所属機関
author={Kosasih, Edward Elson and Joshi, Rucha Bhalchandra and Channegowda, Janamejaya},
journal={arXiv preprint arXiv:2111.09314},
year={2021}
}
投稿日付
2021/11/17
背景
- 電池の経年劣化に伴う放電特性の変化は非線形であり、SOCの予測を難しくしていた。
- これまでのモデルは以下の3つに分類される。
- Partial Differential Equations(PDEs)を用いた物理モデルにより電池のパラメータを計算する
- 特定の充放電サイクルに対応する充電容量変化を計算することにフォーカスした現象論的アプローチ
- データ科学的アプローチ。複雑な電気化学モデルを使用しない
新規性
- GNNモデルを時系列データに応用したGTSを電池のデータに適用したモデルGAETSを提案。測定データ(電圧、電流など)間の関係をグラフで表現し、時系列データの予測を行う。
手法
-
電池データ(電圧、電流、充電容量、放電容量、充電エネルギー、放電エネルギー)を使用
-
ある時刻tからT時間分のデータを用いて、その後の所定の時間のデータを予測する。
-
隣接行列をベルヌーイ分布の確率変数で表現し、タスクの一部として学習させる。サンプリングにより勾配情報が流れないため、reparametrization trickで対処している。隣接行列はパラメータ間の相互作用を表すための行列になっている。これを学習させることでパラメータ間の依存関係に関する情報が得られる。
-
データの変換にはGRUを使用。
-
ロス関数は、次の時系列に対する予測誤差に加えて、オートエンコーダーのロスを加えている。これは正則化の効果がある。
結果
既存モデルより精度向上。人間でも予測できそうなパターンな気もするが。

参考文献
[2022] A Hybrid Machine Learning Approach for Structure Stability Prediction in Molecular Co-crystal Screenings
概要
DFT計算値とベースラインの物理モデルとの差分を機械学習ポテンシャルで予測するΔ-MLを共結晶に適用。ベースラインモデルよりもForceの予測誤差が1/10になり、構造緩和の精度が向上した。
論文リンク
https://pubs.acs.org/doi/10.1021/acs.jctc.2c00343
著者/所属機関
投稿日付
2022/6/16
背景
-
共結晶は分子の配列により特性が決まるが、膨大な組み合わせがあるため、高速に予測する手法が必要。
-
共結晶の構造安定性は比較的弱い長距離相互作用により繊細に決まっており、適切にモデルに取り込む必要がある。
-
Δ-MLは近距離相互作用をMLで取り扱い、長距離相互作用のベースラインと組み合わせることで精度の向上に成功させている。
新規性
- ΔーMLを共結晶に適用
手法
-
長距離相互作用を取り扱う物理モデルベースラインと近距離相互作用を取り扱うMLモデルを組み合わせる。
-
MLモデルを分子内、分子間の寄与に分解する。
-
ベースラインとして、分散力補正をしたDFTBを用いた。MLモデルとしてはSOAP特徴量を用いたGAPを構築。分子間、分子内のエネルギーをそれぞれモデリング。全エネルギーは以下。

-
教師データとしてはPBE0+MBDで計算した値を使用。分子結晶の安定性評価時にはMBDの寄与が重要であることが知られている。
-
計算対象の構造は以下のように取得。
- PyXtalパッケージを用いてまず10000構造作成。
- DFTB+D4で構造緩和し、farthest point sampling(ref 1)で構造をサンプリング。SOAP特徴量で構造の類似度を評価。最も類似度が低い構造をsequentialに追加する。合計1000構造のデータセットを作成
- 実験で知られている77構造に対し、ランダムに変位させた構造を追加。
結果
- DFTB+D4とΔ-GAPの精度は以下。ΔーGAPによりエネルギーの分散が大きく下がっており、構造間の相対比較の精度が向上している。ForceのMAEはΔ-GAPにより大きく改善した。(324 -> 37 meV/Å)

- PBE+MBD, DFTB+D4, Δ-GAPで構造緩和した構造に対し、PBE0+MBDで構造緩和した構造との差を比較した。

RMSDではPBE+MBDがΔ-GAPよりも良い結果であったが、1000倍くらいの計算コストがかかるので、計算コストを考えるとΔ-GAPが優れている。
参考文献
farthest point sampling:
Ceriotti, M.; Willatt, M. J.; Csányi, G. Machine Learning of Atomic-Scale Properties Based on Physical Principles. Handb. Mater. Model. 2018, 1−27.
[2022] GeoDiff a Geometric Diffusion Model for Molecular Conformation Generation
概要
分子のコンフォメーション予測。拡散モデルを使用した生成モデルにおいて、SE(3)同変なマルコフ過程を構築することで、原子座標の直接生成が可能になった。特に大きな分子において、他の手法よりも高精度にコンフォメーションを予測可能。

論文リンク
https://arxiv.org/abs/2203.02923
著者/所属機関
@Article{xu2022geodiff,
title={Geodiff: A geometric diffusion model for molecular conformation generation},
author={Xu, Minkai and Yu, Lantao and Song, Yang and Shi, Chence and Ermon, Stefano and Tang, Jian},
journal={arXiv preprint arXiv:2203.02923},
year={2022}
}
投稿日付
2022/3/6
背景
-
MDやMCMCでのコンフォメーション予測は、特に大きな分子において計算コストが高かった。
-
コンフォメーション予測は、並進や回転に対し不変であるという要請から、これまでの深層学習による予測は、原子間距離マトリックスを予測するなど、原子座標を直接予測するものではなかった(座標を直接予測するCVGAEという手法もあるが、並進、回転に対し不変ではなく精度が悪かった)。これはマトリックスから構造を予測する際に誤差が蓄積し、精度が悪くなる傾向がある。そのため並進、回転に対し不変であることを考慮した原子座標の予測が望ましい。
-
安定状態から熱的に拡散し非平衡状態になる過程から着想し、分子構造の予測を行うGEODIFFを開発。これは並進、回転不変であるという要請を満たす。
新規性
- ノイズ除去拡散モデルのマルコフ過程を並進、回転不変な分布と同変なカーネルを用いて構築した。これにより、原子座標を直接予測することが可能であり、結合の集合から構造に変換するときに生じる誤差などをなくすことが可能である。多峰性の高い分布において精度が高く、高品質で多様性のある構造を生成することができる。
手法
-
ノイズの分布から分子のコンフォメーションに変換する過程をマルコフ過程として学習する。変換のカーネルが学習できれば良い。
-
安定な配置から、各時刻で統計的なノイズが原子位置に付与される拡散過程の逆を学習。
-
拡散過程では以下のように事後分布を設定する。

このとき、不変性は課してはいない。時刻Tでは、座標の確率分布は標準正規分布になる。 -
生成過程では拡散過程の逆を辿り、ある分子グラフの条件のもとで、ノイズからコンフォメーションを生成する。学習可能なパラメータを含む、条件付きマルコフ過程で定式化。

μθは平均を予測するためのネットワークになっている。またpθ(C0)では並進、回転に対し不変な確率分布である必要がある。 -
マルコフ過程のカーネルを並進、回転に対し同変にすることで、不変な確率密度に対し変換を繰り返した、最終の確率分布は不変になる。そのため、不変な確率分布(生成モデルの入力になる分布)と同変なカーネルを設計すれば良い。
-
不変な初期分布は以下を参考にしたとのこと。
Jonas Köhler, Leon Klein, and Frank Noe. Equivariant flows: Exact likelihood generative learning for symmetric densities. In Proceedings of the 37th International Conference on Machine Learning, 2020.
これによると並進不変が実現できる。等方的な正規分布であれば回転不変はそもそも実現できている。 -
同変なカーネルはgraph field network(GFN)というものを使用。

-
学習は変分下限の最大化で実施
結果

-
特に大きな分子において、他の手法よりも優れていた。
-
多峰性分布において正確かつ多様な構造の生成が可能であった。
参考文献
[2022] Xtal2DoS: Attention-based Crystal to Sequence Learning for Density of States Prediction
概要
論文リンク
著者/所属機関
投稿日付
背景
結晶構造を対象にした機械学習はスカラー量の例が多く、DoS(phonon density of states: phDoS, electronic density of states: eDoS)などのスペクトルに関しての予測例が少ない。
単純にスペクトルを構成するスカラー量を一度に出力すると、連続的であるという要請を満たさない。Mat2Specはこの問題に対し対照学習、VAEを使用することで連続量の相関を扱っている。
新規性
transformer型のアーキテクチャを用いてDoSを予測するXtal2DoSを提案。
手法
モデル
ノードの特徴量としてCGCNNのembeddinbgを使用。
attention部分は、ノード特徴量をq,k,vに変換する。このとき、kはエッジを変換したものも加えることでエッジの情報を取り込んだattention weightを出力する。
さらに残差接続を加え、元のノード特徴量との線形和をとることでノード特徴量の更新を行う。
最終層のノード特徴量の平均をとり、global featureとした。これを初期のmemory入力とし、以下の4種のモデルを検証した。
- RNN
- Chunk RNN
- Chunk RNN + Attention
- Transformer
RNNでは127個のスカラーデータを順番に出力し、DoSとする。Chunk RNNでは分割したDoSデータを順番に出力。これにattentionを加えたモデルも作成。
transformerのモデルでは
結果
参考文献
Recommend Projects
-
 React
React
A declarative, efficient, and flexible JavaScript library for building user interfaces.
-
Vue.js
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
-
 Typescript
Typescript
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
-
TensorFlow
An Open Source Machine Learning Framework for Everyone
-
Django
The Web framework for perfectionists with deadlines.
-
 Laravel
Laravel
A PHP framework for web artisans
-
D3
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
-
Recommend Topics
-
javascript
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
-
web
Some thing interesting about web. New door for the world.
-
server
A server is a program made to process requests and deliver data to clients.
-
Machine learning
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
-
Visualization
Some thing interesting about visualization, use data art
-
Game
Some thing interesting about game, make everyone happy.
Recommend Org
-
Facebook
We are working to build community through open source technology. NB: members must have two-factor auth.
-
Microsoft
Open source projects and samples from Microsoft.
-
Google
Google ❤️ Open Source for everyone.
-
Alibaba
Alibaba Open Source for everyone
-
D3
Data-Driven Documents codes.
-
Tencent
China tencent open source team.