
![]()



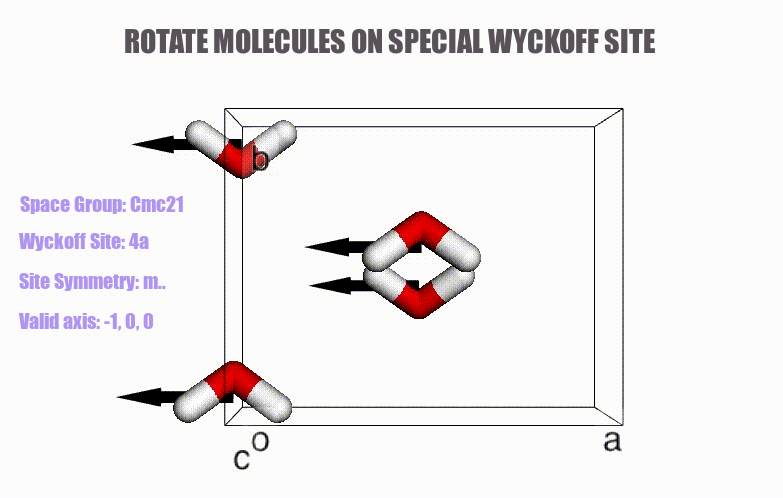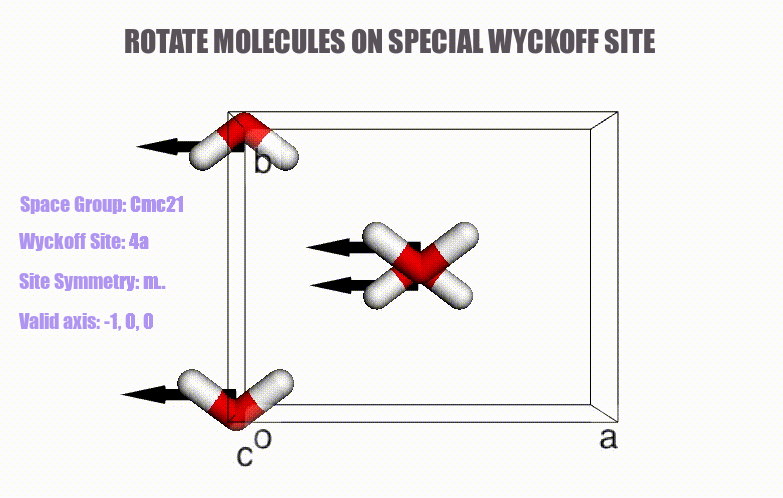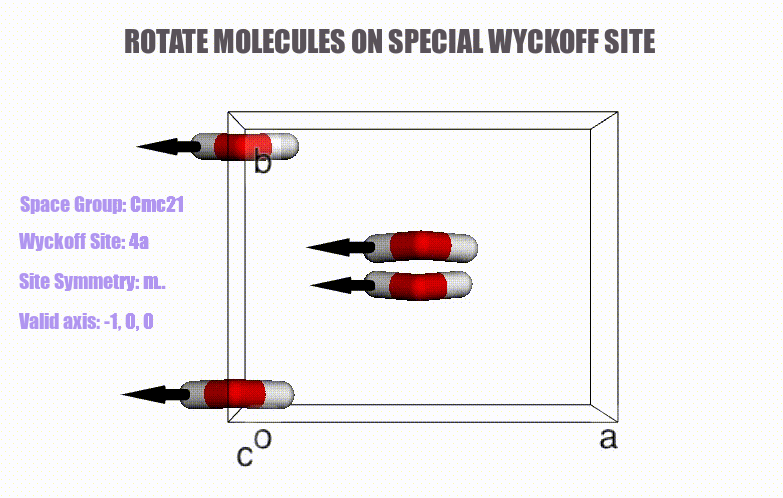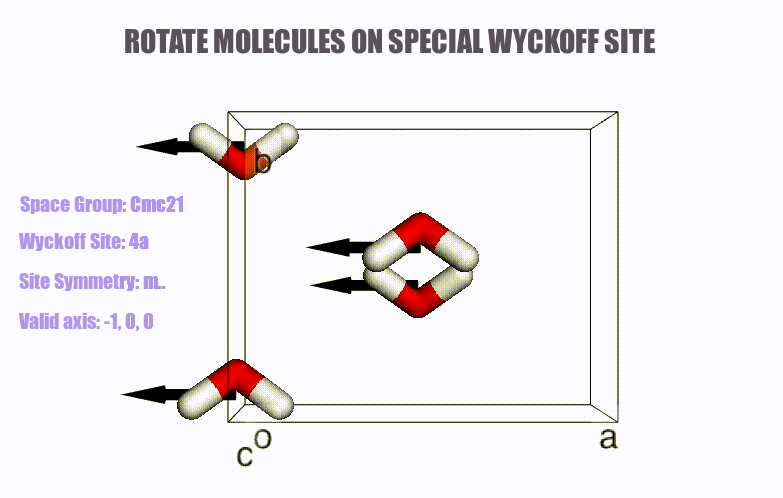
PyXtal is an open source Python package which was initiated by Qiang Zhu and Scott Fredericks at department of Physics and Astronomy, University of Nevada Las Vegas. The goal of PyXtal is to develop a fundamental library to allow one to design the material structure with a certain symmetry constraint. These structures can exported to various structural formats for further study. See the documentation for more information.
To contribute to this project, please check How to contribute?.
- Generation of atomic structures for a given symmetry and stoichiometry (0-3D)
- Generation of molecular crystals (1-3D) with the support of special Wyckoff positions.
- Structure manipulation via subgroup/supergroup symmetry relation
- Geometry optimization from built-in and external optimization methods.
- Internal support of
ciffile and many other formats viapymatgenorASE. - Easy access to symmetry information (e.g., Wyckoff, site symmetry and international symbols).
- X-ray diffraction analysis and its online application
To install the code, one just needs to do
pip install pyxtalor
pip install --upgrade git+https://github.com/qzhu2017/PyXtal.git@masterFredericks S, Parrish K, Sayre D, Zhu Q*(2020) PyXtal: a Python Library for Crystal Structure Generation and Symmetry Analysis. [arXiv link]
@article{pyxtal,
title = "PyXtal: A Python library for crystal structure generation and symmetry analysis",
journal = "Computer Physics Communications",
volume = "261",
pages = "107810",
year = "2021",
issn = "0010-4655",
doi = "https://doi.org/10.1016/j.cpc.2020.107810",
url = "http://www.sciencedirect.com/science/article/pii/S0010465520304057",
author = "Scott Fredericks and Kevin Parrish and Dean Sayre and Qiang Zhu",
}This is an open-source project. Its growth depends on the community. To contribute to PyXtal, you don't necessarily have to write the code. Any contributions from the following list will be helpful.
the PyXtal project and recommend it to your colleagues/friends
- Open an
to report the bug or address your wishlist or improve our documentation
the repository and send us the pull request





![dependabot[bot] avatar](https://avatars.githubusercontent.com/in/29110?v=4 "dependabot[bot]")























































































































