An R package for the estimation and removal of cell free mRNA contamination in droplet based single cell RNA-seq data.
The problem this package attempts to solve is that all droplet based single cell RNA-seq experiments also capture ambient mRNAs present in the input solution along with cell specific mRNAs of interest. This contamination is ubiquitous and can vary hugely between experiments (2% - 50%), although around 10% seems reasonably common.
There's no way to know in advance what the contamination is in an experiment, although solid tumours and low-viability cells tend to produce higher contamination fractions. As the source of the contaminating mRNAs is lysed cells in the input solution, the profile of the contamination is experiment specific and produces a batch effect.
Even if you decide you don't want to use the SoupX correction methods for whatever reason, you should at least want to know how contaminated your data are.
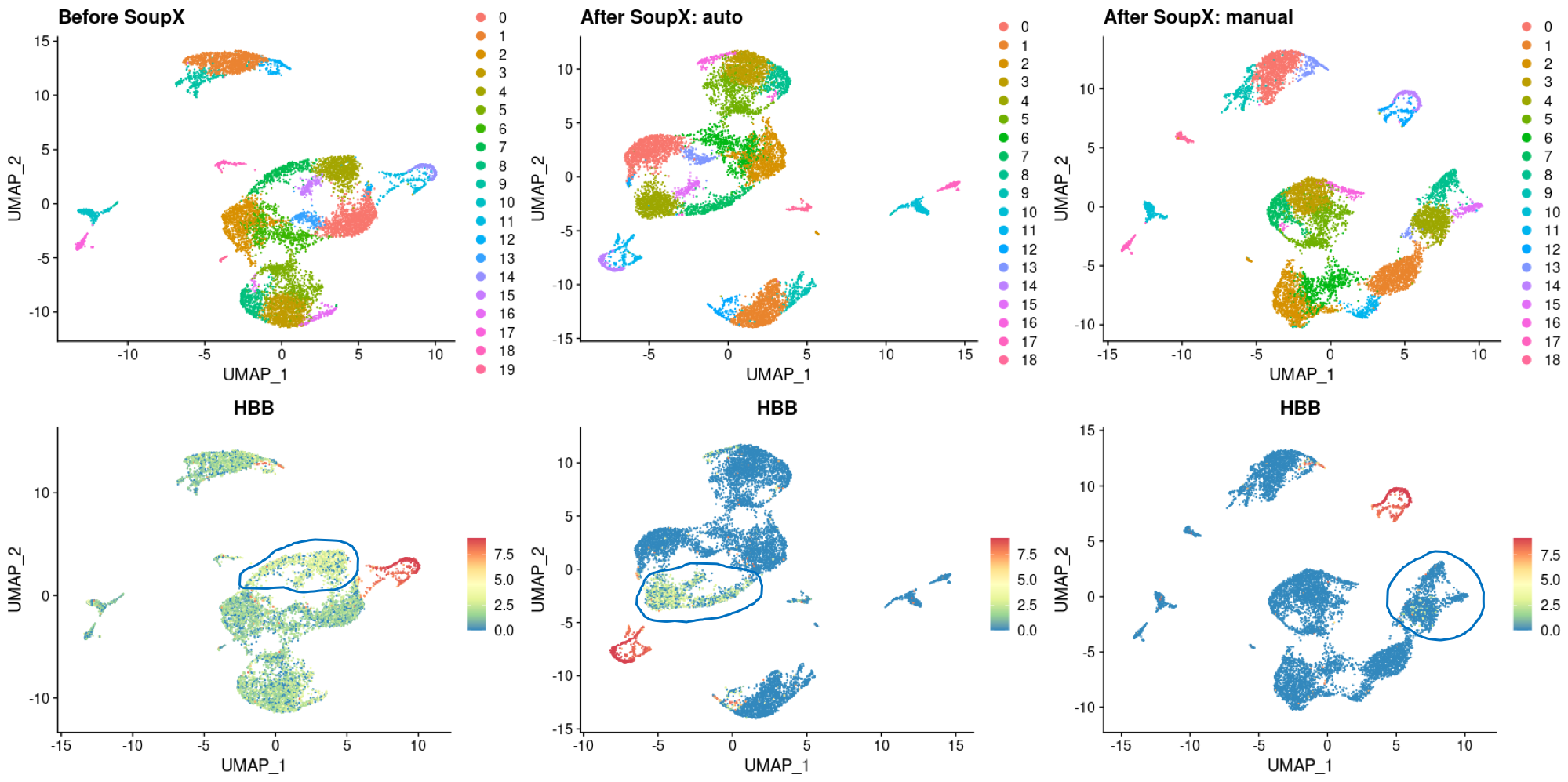
NOTE: From v1.3.0 onward SoupX now includes an option to automatically estimate the contamination fraction. It is anticipated that this will be the preferred way of using the method for the vast majority of users. This function (autoEstCont) depends on clustering information being provided. If you are using 10X data mapped with cellranger, this will be loaded automatically, but otherwise it must be provided explicitly by the user using setClusters.
The latest stable release can be installed from CRAN in the usual way by running,
install.packages('SoupX')If you want to use the latest development version, install it by running,
devtools::install_github("constantAmateur/SoupX",ref='devel')Finally, if you want to use the per-cell contamination estimation (which you almost certainly won't need to), install the branch STAN
devtools::install_github("constantAmateur/SoupX",ref='STAN')If you encounter errors saying multtest is unavailable, please install this manually from bioconductor with:
BiocManager::install('multtest')Decontaminate one channel of 10X data mapped with cellranger by running:
sc = load10X('path/to/your/cellranger/outs/folder')
sc = autoEstCont(sc)
out = adjustCounts(sc)or to manually load decontaminate any other data
sc = SoupChannel(table_of_droplets,table_of_counts)
sc = setClusters(sc,cluster_labels)
sc = autoEstCont(sc)
out = adjustCounts(sc)out will then contain a corrected matrix to be used in place of the original table of counts in downstream analyses.
The methodology implemented in this package is explained in detail in this paper.
A detailed vignette is provided with the package and can be viewed here.
If you use SoupX in your work, please cite: "Young, M.D., Behjati, S. (2020). SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data, GigaScience, Volume 9, Issue 12, December 2020, giaa151bioRxiv, 303727, https://doi.org/10.1093/gigascience/giaa151"
The automatic estimation of the contamination implemented in autoEstCont makes the assumption that there is sufficient diversity in the raw data to identify marker genes (as such genes are commonly useful for estimating the contamination). If your data is either extremely homogenous (i.e., all one cell type, for example a cell line) or your number of cells is very low (a few hundred or less), then this assumption is unlikely to hold. In such situations you should think hard about if you really want to include data with such severe limitations. But if you're sure you do, the best approach is probably to manually specify a contamination fraction in line with what you would expect from similar experiments.
The first thing to do is check that you are providing clustering information, either by doing clustering yourself and running setClusters before adjustCounts or by loading it automatically from load10X. Cluster information allows far more contamination to be identified and safely removed.
The second thing to consider is if the contamination rate estimate looks plausible. As estimating the contamination rate is the part of the method that requires the most user input, it can be prone to errors. Generally a contamination rate of 2% or less is low, 5% is usual, 10% moderate and 20% or above very high. Of course your experience may vary and these expectations are based on fresh tissue experiments on the 10X 3' platform.
Finally, note that SoupX has been designed to try and err on the side of not throwing out real counts. In some cases it is more important to remove contamination than be sure you've retained all the true counts. This is particularly true as "over-removal" will not remove all the expression from a truly expressed gene unless you set the over-removal to something extreme. If this describes your situation you may want to try manually increasing the contamination rate by setting setContaminationFraction and seeing if this improves your results.
Generally the gene sets that work best are sets of genes highly specific to a cell type that is present in your data at low frequency. Think HB genes and erythrocytes, IG genes and B-cells, TPSB2/TPSAB1 and Mast cells, etc. Before trying anything more esoteric, it is usually a good idea to at least try out the most commonly successful gene sets, particularly HB genes. If this fails, the plotMarkerDistribution function can be used to get further inspiration as described in the vignette. If all of this yields nothing, we suggest trying a range of corrections to see what effect this has on your downstream analysis. In our experience most experiments have somewhere between 2-10% contamination.
At this point we assume that you have chosen a set (or sets) of genes to use to estimate the contamination. The default behaviour (with 10X data) is to look for cells with strong evidence of endogenous expression of these gene sets in all cells, then exclude any cluster with a cell that has strong evidence of endogenous expression. This conservative behaviour is designed to stop the over-estimation of the contamination fraction, but can sometimes make estimation difficult. If all clusters have at least one cell that "looks bad" you have 3 options.
- Recluster the data to produce more clusters with fewer cells per cluster. This is the preferred option, but requires more work on the users part.
- Make the criteria for declaring a cell to be genuinely expressing a gene set less strict. This seldom works, as usually when a cell is over the threshold, it's over by a lot. But in some cases tweaking the values
maximumContaminationand/orpCutcan yield usable results. - Set
clusters=FALSEto forceestimateNonExpressingCellsto consider each cell independently. If you are going to do this, it is worth making the criteria for excluding a cell more permissive by decreasingmaximumContaminationas much as is reasonable.
- Added some checks and security around setting of clusters with
setClusters. - Add warnings and extra documentation about library complexity to
autoEstCont. - Fix pointSize bug in plotting function.
- Merge pull adding support for multi.
load10X now requires the version of Seurat::Read10X that does not strip out the numeric suffix.
First CRAN version of the code. The one significant change other than tweaks to reach CRAN compatibility is that the correction algorithm has been made about 20 times faster. As such, the parallel option was no longer needed and has been removed. Also includes some other minor tweaks.
Addition of autoEstCont function to automatically estimate the contamination fraction without the need to specify a set of genes to use for estimation. A number of other tweaks and bug fixes.
Some bug fixes from v1.0.0. Added some helper functions for integrating metadata into SoupChannel object. Further integration of cluster information in estimation of contamination and calculation of adjusted counts. Make the adjustCounts routine parallel.
Review of method, with focus on simplification of code. Functions that were being used to "automate" selection of genes for contamination estimation have been removed as they were being misused. Clustering is now used to guide selection of cells where a set of genes is not expressed. Default now set to use global estimation of rho. A hierarchical bayes routine has been added to share information between cells when the user does use cell specific estimation. See NOTE for further details.
Now passes R CMD check without warnings or errors. Added extra vignette on estimating contamination correctly. Changed the arguments for the interpolateCellContamination function and made monotonically decreasing lowess the default interpolation method. A number of other plotting improvements.
Added lowess smoothing to interpolation and made it the default. Modified various functions to allow single channel processing in a more natural way. Some minor bug fixes.
Integrated estimateSoup into class construction to save memory when loading many channels. Added function to use tf-idf to quickly estimate markers. Some minor bug fixes and documentation updates.
Update documentation and modify plot functions to return source data.frame.
A fairly major overhaul of the data structures used by the package. Not compatible with previous versions.
Some bug fixes to plotting routines.
Copyright (c) 2018 Genome Research Ltd.
Author: Matthew Young <[email protected]>
This program is free software: you can redistribute it and/or
modify it under the terms of the GNU General Public License version 3
as published by the Free Software Foundation.
This program is distributed in the hope that it will be useful,
but WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU
General Public License for more details <http://www.gnu.org/licenses/>.























































































































{kind=link}