Originally, CNNC is short for Convolutional neural network co-expression analysis. Co-expression is just one of the tasks CNNC can do, but the name (CNNC) derived from it looks very nice.
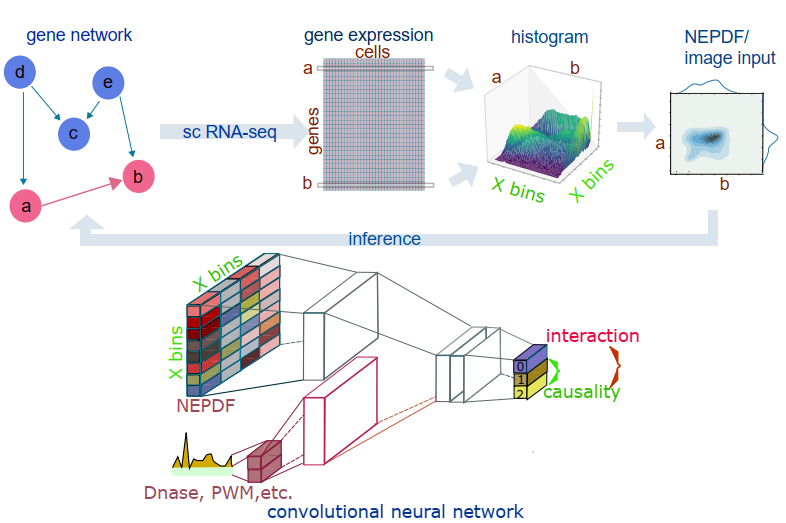
CNNC aims to infer gene-gene relationships using single cell expression data. For each gene pair, sc RNA-Seq expression levels are transformed into 32×32 normalized empirical probability function (NEPDF) matrices. The NEPDF serves as an input to a convolutional neural network (CNN). The intermediate layer of the CNN can be further concatenated with input vectors representing Dnase-seq and PWM data. The output layer can either have a single, three or more values, depending on the application. For example, for causality inference the output layer contains three probability nodes where p0 represents the probability that genes a and b are not interacting, p1 encodes the case that gene a regulates gene b, and p2 is the probability that gene b regulates gene a.
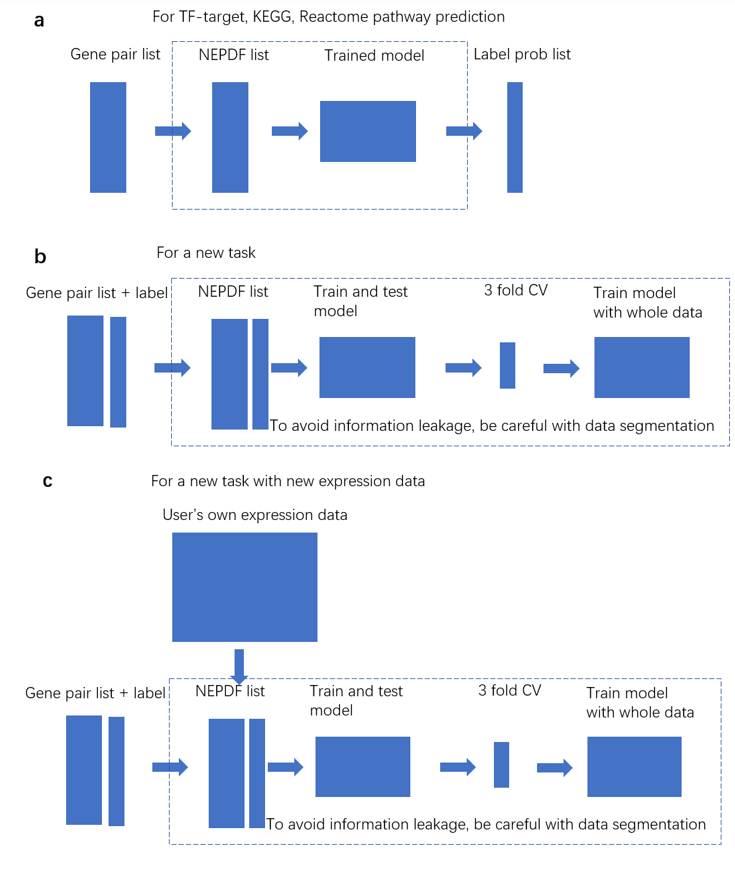
(a) Pipeline for TF-target, KEGG and Reactome edge predictions. Users only need to provide gene-pair candidate list. TF-tatget prediction is cell type specific. Here we provide the model for mESC TF prediction. Please use mESC expression data to generate mESC subset and then do following NEPDF generation and classification of training and test, and use the big scRNA-seq and bulk data to do pathway tasks. (b) Pipeline for a new task with the expression data we collected. Users need to provide gene-pair candidate list to generate NEPDF list and label list to train and test model. (c) Pipeline for a new task with the expression data users collect. Users need to provide gene-pair candidate list, their own expression data to generate NEPDF list, and label list to train and test model.
https://s3.amazonaws.com/mousescexpression/rank_total_gene_rpkm.h5
https://mousescexpression.s3.amazonaws.com/bone_marrow_cell.h5
https://s3.us-east-2.amazonaws.com/mousebulkexprssion/mouse_bulk.h5
https://mousescexpression.s3.amazonaws.com/mesc_cell.h5
https://mousescexpression.s3.amazonaws.com/dendritic_cell.h5
Author's environment is python 3.6.3 in a Linux server which is now running Centos 6.5 as the underlying OS and Rocks 6.1.1 as the cluster management revision.
And Author uses theano as the Keras backend in python.
Author's GPU is GeForce GTX 1080. If the latest theano does not work, please try some older versions.
Although not necessary, we strongly recommend GPU acceleration and conda management for package, dependency and environment to save time. With conda, the total software, package module installation time in Python should be less than one hour.
(see folder for details)
gene_pair_list is the list that contains gene pairs and their labels. format : 'GeneA GeneB ' or 'GeneA GeneB 0'
such as mmukegg_new_new_unique_rand_labelx_sy.txt and mmukegg_new_new_unique_rand_labelx.txt in data folder.
users also need to provide data_separation index_list which is a number list dividing gene_pair_list into small parts.
Here we use data separation index list to divide gene pairs into small data parts, and make sure that the gene pairs in each index inteval is completely isolated from others. We can evaluate CNNC's performance on only a small data part.
If users do not want to specified separate data, they can just generate a index list to divide the data into N equal parts.
python get_xy_label_data_cnn_combine_from_database.py /home/yey/CNNC-master/data/bulk_gene_list.txt /home/yey/CNNC-master/data/sc_gene_list.txt /home/yey/CNNC-master/data/mmukegg_new_new_unique_rand_labelx.txt /home/yey/CNNC-master/data/mmukegg_new_new_unique_rand_labelx_num_sy.txt /home/yey/sc_process_1/new_bulk_mouse/prs_calculation/mouse_bulk.h5 /home/yey/sc_process_1/rank_total_gene_rpkm.h5 0
#################INPUT################################################################################################################################
#1,
bulk_gene_list.txtis the list that converts bulk expression data gene set into gene symbol IDs. Format:'gene symbol IDs\t bulk gene ID'. Set asNoneif you do not have it.
#2,
sc_gene_list.txtis the list that converts sc expression data gene set into gene symbol IDs. Format:'gene symbol IDs\t sc gene ID'.please notice that mesc single cell data is not from the big 40k data, so we proivde a new sc_gene_list.txt file for it. . Set asNoneif you do not have it.
#3,
gene_pair_listis the list that contains gene pairs and their labels. format :'GeneA GeneB'
#4,
data_separation index_listis a number list that divides gene_pair_list into small parts
#Here we use data separation index list to divide gene pairs into small data parts, and make sure that the gene pairs in each index inteval is completely isolated from others. And we can evaluate CNNC's performance on only a small data part.
#if users do not need to separate data, they can just generate a index list to divide the data into N equal parts.
#5,
bulk_expression_datait should be a hdf5 format. users can use their own data or data we provided. Set asNoneif you do not have it.
#6,
sc expression datait should be a hdf5 format. users can use their own data or data we provided. Set asNoneif you do not have it.
#7,
flag, 0 means do not generate label list; 1 means to generate label list.
If user does not have bulk (single cell) data, just put
Nones forbulk_gene_list.txt(sc_gene_list.txt) andbulk_expression_data(sc expression data).
#################OUTPUT
It generates a NEPDF_data folder, and a series of data files containing Nxdata_tf (NEPDF file) and zdata_tf (gene symbol pair file) for each data part divided.
Here we use gene symbol information to align bulk, scRNA-seq and gene pair's gene sets. In our own data, scRNA-seq used entrez ID, bulk RNA-seq used ensembl ID, gene pair list used gene symbol ID, thus we used bulk_gene_list.txt and sc_gene_list.txt to convert all the IDs to gene symbols. Please also do IDs convertion for bulk and scRNA-seq data if users want to use their own expression data.
python predict_no_y.py 9 /home/yey/CNNC-master/NEPDF_data 3 /home/yey/CNNC-master/trained_models/KEGG_keras_cnn_trained_model_shallow.h5
(In the models folder are trained models for KEGG and Reactome database respectively)
gene_pair_list is the list that contains gene pairs and their labels. format : 'GeneA GeneB 0'
such as mmukegg_new_new_unique_rand_labelx_sy.txt and mmukegg_new_new_unique_rand_labelx.txt in data folder.
users also need to provide data_separation index_list which is a number list dividing gene_pair_list into small parts
Here we use data separation index list to divide gene pairs into small data parts, and make sure that the gene pairs in each index inteval is completely isolated from others. And we can evaluate CNNC's performance on only a small data part.
If users do not need to separate data, they can just generate a index list to divide the data into N equal parts.
python get_xy_label_data_cnn_combine_from_database.py /home/yey/CNNC-master/data/bulk_gene_list.txt /home/yey/CNNC-master/data/sc_gene_list.txt /home/yey/CNNC-master/data/mmukegg_new_new_unique_rand_labelx.txt /home/yey/CNNC-master/data/mmukegg_new_new_unique_rand_labelx_num_sy.txt /home/yey/sc_process_1/new_bulk_mouse/prs_calculation/mouse_bulk.h5 /home/yey/sc_process_1/rank_total_gene_rpkm.h5 1
#################INPUT################################################################################################################################
#1,
bulk_gene_list.txtis the list that converts bulk expression data gene set into gene symbol IDs. Format:'gene symbol IDs\t bulk gene ID'. Set asNoneif you do not have it.
#2,
sc_gene_list.txtis the list that converts sc expression data gene set into gene symbol IDs. Format:'gene symbol IDs\t sc gene ID'. Set asNoneif you do not have it.
#3,
gene_pair_listis the list that contains gene pairs and their labels. format :'GeneA GeneB 0'
#4,
data_separation_index_listis a number list that divide gene_pair_list into small parts
#Here we use data separation index list to divide gene pairs into small data parts, and make sure that the gene pairs in each index inteval is completely isolated from others. And we can evaluate CNNC's performance on only a small data part.
#if users do not want to separate data, they can just generate a index list to divide the data into N equal parts.
#5,
bulk_expression_datait should be a hdf5 format. users can use their own data or data we provided. Set asNoneif you do not have it.
#6,
sc_expression_datait should be a hdf5 format. users can use their own data or data we provided. Set asNoneif you do not have it.
#7,
flag, 0 means do not generate label list; 1 means to generate label list.
If user does not have bulk (single cell) data, just put
Nones forbulk_gene_list.txt(sc_gene_list.txt) andbulk_expression_data(sc expression data). #################OUTPUT
It generate a NEPDF_data folder, and a series of data files containing Nxdata_tf (NEPDF file), ydata_tf (label file) and zdata_tf (gene symbol pair file) for each data part divided.
module load cuda-8.0 (it is to use GPU)
srun -p gpu --gres=gpu:1 -c 2 --mem=20Gb python train_with_labels_three_foldx.py 9 /home/yey/CNNC-master/NEPDF_data 3 > results.txt
#######################OUTPUT
It generates three cross_Validation folder whose name begins with YYYYY, in which keras_cnn_trained_model_shallow.h5 is the trained model






















































































