

(Pronounced "churro.")
Lots of tools for analyzing " 'omic" datasets can produce feature rankings. These rankings can be used as a guide to look at the log-ratios of certain features in a dataset. Qurro is a tool for visualizing and exploring both of these types of data.
The term "feature rankings" includes differentials, which we define as the estimated log-fold changes for features' abundances across different sample types. You can get this sort of output from lots of "differential abundance" tools, including but definitely not limited to ALDEx2, Songbird, Corncob, DESeq2, edgeR, etc.
The term "feature rankings" also includes feature loadings in a biplot (see Aitchison and Greenacre 2002); you can get biplots from running DEICODE, which is a tool that works well with microbiome datasets, or from a variety of other methods.
Differentials and feature loadings alike can be interpreted as rankings -- you can sort them numerically to "rank" features based on their association with some sort of variation in your dataset.
A common use of these rankings is examining the log-ratios of particularly high- or low-ranked features across the samples in your dataset, and seeing how these log-ratios relate to your sample metadata (e.g. "does this log-ratio differ between 'healthy' and 'sick' samples?"). For details as to why this approach is useful, check out this open access paper.
Qurro is an interactive web application for visualizing feature rankings and log-ratios. It does this using a two-plot interface: on the left side of the screen, a "rank plot" shows how features are ranked for a selected ranking, and on the right side of the screen a "sample plot" shows the log-ratios of selected features' abundances within samples. There are a variety of controls available for selecting features for a log-ratio, and changing the selected log-ratio updates both the rank plot (highlighting selected features) and the sample plot (changing the y-axis value of each sample to match the selected log-ratio).
A paper describing Qurro is now available at NAR Genomics and Bioinformatics here.
Qurro can be used standalone (as a Python 3 script that generates a
folder containing a HTML/JS/CSS visualization) or as a
QIIME 2 plugin (that generates a QZV file that can be
visualized at view.qiime2.org or by using
qiime tools view).
Qurro is still being developed, so backwards-incompatible changes might occur. If you have any bug reports, feature requests, questions, or if you just want to yell at me, then feel free to open an issue in this repository!
See the Qurro website for a list of interactive demos using real datasets.
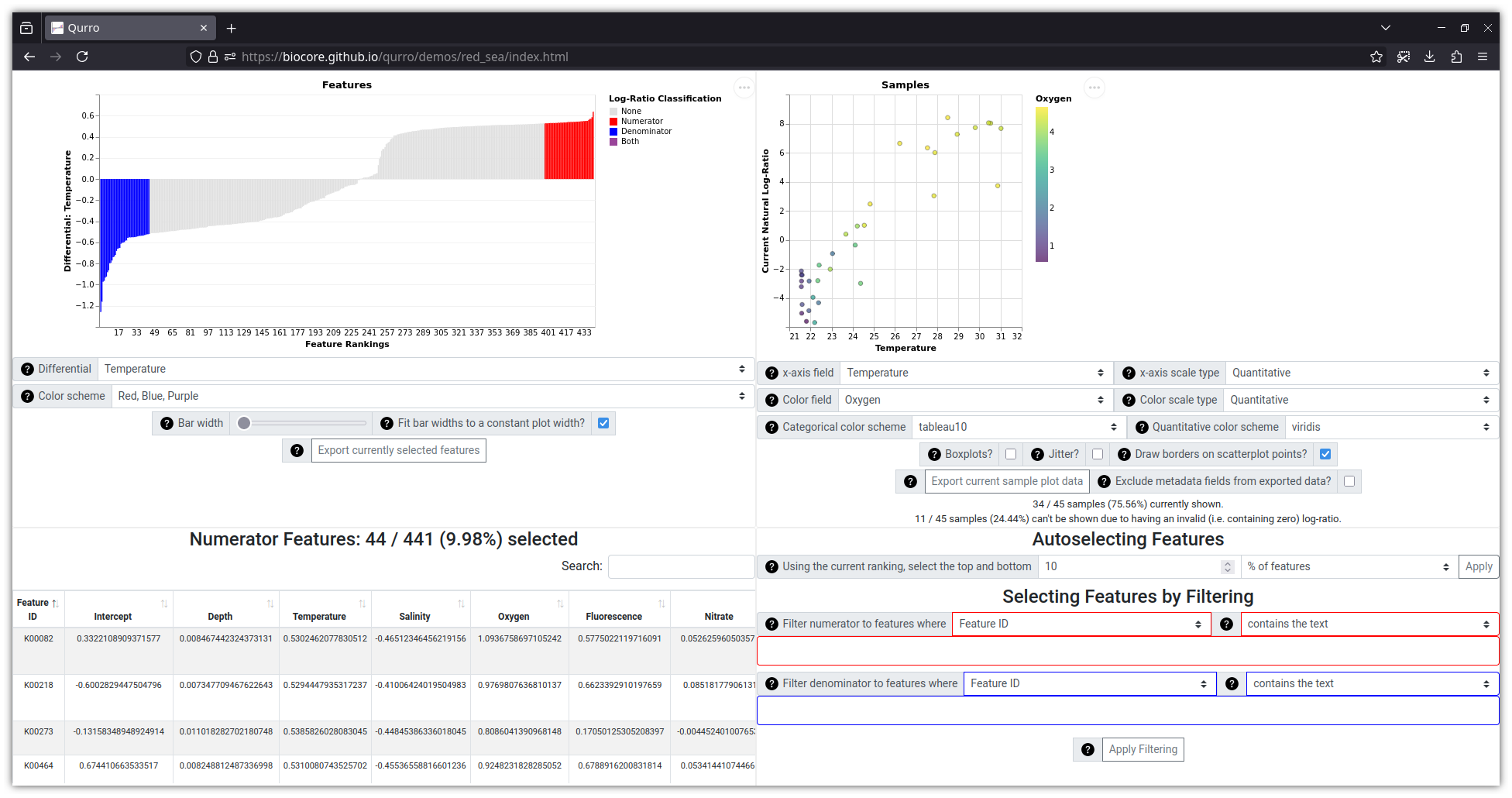
This visualization (which uses data from this study, with differentials generated by Songbird) can be viewed online here.
You can install Qurro using pip or conda.
If you're using Qurro within QIIME 2, you will need a QIIME 2 version of at least 2020.11.
If you're using Qurro outside of QIIME 2, you will need a Python version of at least 3.6 and less than 3.10.
In either case, Qurro should work with most modern web browsers; Firefox or Chrome are recommended.
pip install cython "numpy >= 1.12.0"
pip install qurroconda install -c conda-forge qurroCertain characters in column names in the sample metadata, feature metadata (if passed), and feature differentials (if passed) will be replaced with similar characters or just removed entirely:
| Old Character(s) | New Character |
|---|---|
. |
: |
] |
) |
[ |
( |
\ |
| |
' or " |
Nothing |
This is due to some downstream issues with handling these sorts of characters in field names. See this issue for context.
These tutorials are all good places to start, depending on what sort of data and feature rankings you have.
-
- Data Summary: Color composition data from abstract paintings
- Feature rankings: Feature loadings in an arbitrary compositional biplot
- Qurro used through QIIME 2 or standalone?: Standalone
-
- Data Summary: Microbiome 16S rRNA marker gene sequencing data from four types of body site samples
- Feature rankings: Feature loadings in a DEICODE biplot
- Qurro used through QIIME 2 or standalone?: QIIME 2
-
- Data Summary: Gene expression ("RNA-Seq") data from TCGA tumor and "solid tissue normal" samples
- Feature rankings: ALDEx2 differentials
- Qurro used through QIIME 2 or standalone?: Standalone
There are a lot of different ways to select features in Qurro, and the interface can be difficult to get used to. This document describes all of these methods, and provides some examples of where they could be useful in practice.
These tutorials show examples of using Qurro in identical ways both inside and outside of QIIME 2.
Qarcoal (pronounced "charcoal") is a new part of Qurro that lets you compute log-ratios based on taxonomic searching directly from the command-line. This can be useful for a variety of reasons.
Currently, Qarcoal is only available through Qurro's QIIME 2 plugin interface.
Please see qarcoal_example.ipynb
for a demonstration of using Qarcoal.
We presented this poster on Qurro at the 2019 CRISP Annual Review. The data shown here is already slightly outdated compared to the actual Qurro paper (e.g. the differentials are slightly different), but feel free to check out the poster anyway!
Code files for the following projects are distributed within
qurro/support_files/vendor/.
See the qurro/dependency_licenses/ directory for copies of these software projects'
licenses (each of which includes a respective copyright notice).
- Vega
- Vega-Lite
- Vega-Embed
- jQuery
- DataTables
- RequireJS
- Bootstrap
- Bootstrap Icons
- We make use of the "Question fill" icon's SVG, as well as some example code for embedding this or other icons in CSS.
- Popper.js (included within the Bootstrap JS "bundle" file)
The following software projects are required for Qurro's python code to function, although they are not distributed with Qurro (and are instead installed alongside Qurro).
For python testing/style checking, Qurro uses
pytest,
pytest-cov,
flake8, and
black. You'll also need to have QIIME 2
installed to run most of the python tests (note that, due to click vs. black
vs. QIIME 2 dependency issues, you should use a QIIME 2 environment of at least
2022.8; see
CONTRIBUTING.md
for details).
For JavaScript testing/style checking, Qurro uses Mocha, Chai, mocha-headless-chrome, nyc, jshint, and prettier.
Qurro also uses GitHub Actions and Codecov.
The Jupyter notebooks in Qurro's example_notebooks/ folder are automatically
rerun using nbconvert,
also.
The test data located in qurro/tests/input/mackerel/ were exported from
QIIME 2 artifacts in this repository. These data are from Minich et al. 2020 [1].
The test data located in qurro/tests/input/byrd/ are from
this repository.
These data, in turn, originate from Byrd et al.'s 2017 study on atopic
dermatitis [2].
The test data located in qurro/tests/input/sleep_apnea/
(and in example_notebooks/DEICODE_sleep_apnea/input/)
are from this Qiita study,
which is associated with Tripathi et al.'s 2018 study on sleep apnea [4].
The test data located in qurro/tests/input/moving_pictures/ (and in
example_notebooks/moving_pictures/data/)
are from the QIIME 2 moving pictures tutorial.
The ordination files in these folders were computed based on the
DEICODE moving pictures tutorial.
These data (sans the DEICODE ordination) are associated with Caporaso et al. 2011 [5].
Lastly, the data located in qurro/tests/input/red_sea
(and in example_notebooks/songbird_red_sea/input/, and shown in the
screenshot above) were taken from Songbird's GitHub repository in its
data/redsea/
folder, and are associated with Thompson et al. 2017 [3].
Qurro's logo was created using the Lalezar font. Also, shout out to this gist for showing how to center images in GitHub markdown files (which is more of a hassle than it sounds).
The design of Qurro was strongly inspired by EMPeror and q2-emperor, along with DEICODE. A big shoutout to Yoshiki Vázquez-Baeza for his help in planning this project, as well as to Cameron Martino for a ton of work on getting the code in a distributable state (and making it work with QIIME 2). Thanks also to Jamie Morton, who wrote the original code for producing rank and sample plots from which this is derived.
And thanks to a bunch of the Knight Lab for helping name the tool :)
If you use Qurro in your research, please cite it! The preferred citation for Qurro is this manuscript at NAR Genomics and Bioinformatics. Here's the BibTeX:
@article {fedarko2020,
author = {Fedarko, Marcus W and Martino, Cameron and Morton, James T and González, Antonio and Rahman, Gibraan and Marotz, Clarisse A and Minich, Jeremiah J and Allen, Eric E and Knight, Rob},
title = "{Visualizing ’omic feature rankings and log-ratios using Qurro}",
journal = {NAR Genomics and Bioinformatics},
volume = {2},
number = {2},
year = {2020},
month = {04},
issn = {2631-9268},
doi = {10.1093/nargab/lqaa023},
url = {https://doi.org/10.1093/nargab/lqaa023},
note = {lqaa023},
eprint = {https://academic.oup.com/nargab/article-pdf/2/2/lqaa023/33137933/lqaa023.pdf},
}
[1] Minich, J. J., Petrus, S., Michael, J. D., Michael, T. P., Knight, R., & Allen, E. E. (2020). Temporal, environmental, and biological drivers of the mucosal microbiome in a wild marine fish, Scomber japonicus. mSphere, 5(3), e00401-20. Link.
[2] Byrd, A. L., Deming, C., Cassidy, S. K., Harrison, O. J., Ng, W. I., Conlan, S., ... & NISC Comparative Sequencing Program. (2017). Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Science Translational Medicine, 9(397), eaal4651. Link.
[3] Thompson, L. R., Williams, G. J., Haroon, M. F., Shibl, A., Larsen, P., Shorenstein, J., ... & Stingl, U. (2017). Metagenomic covariation along densely sampled environmental gradients in the Red Sea. The ISME Journal, 11(1), 138. Link.
[4] Tripathi, A., Melnik, A. V., Xue, J., Poulsen, O., Meehan, M. J., Humphrey, G., ... & Haddad, G. (2018). Intermittent hypoxia and hypercapnia, a hallmark of obstructive sleep apnea, alters the gut microbiome and metabolome. mSystems, 3(3), e00020-18. Link.
[5] Caporaso, J. G., Lauber, C. L., Costello, E. K., Berg-Lyons, D., Gonzalez, A., Stombaugh, J., ... & Gordon, J. I. (2011). Moving pictures of the human microbiome. Genome Biology, 12(5), R50. Link.
This tool is licensed under the BSD 3-clause license. Our particular version of the license is based on scikit-bio's license.



































