





If you would like to get started using PHATE, check out the following tutorials.
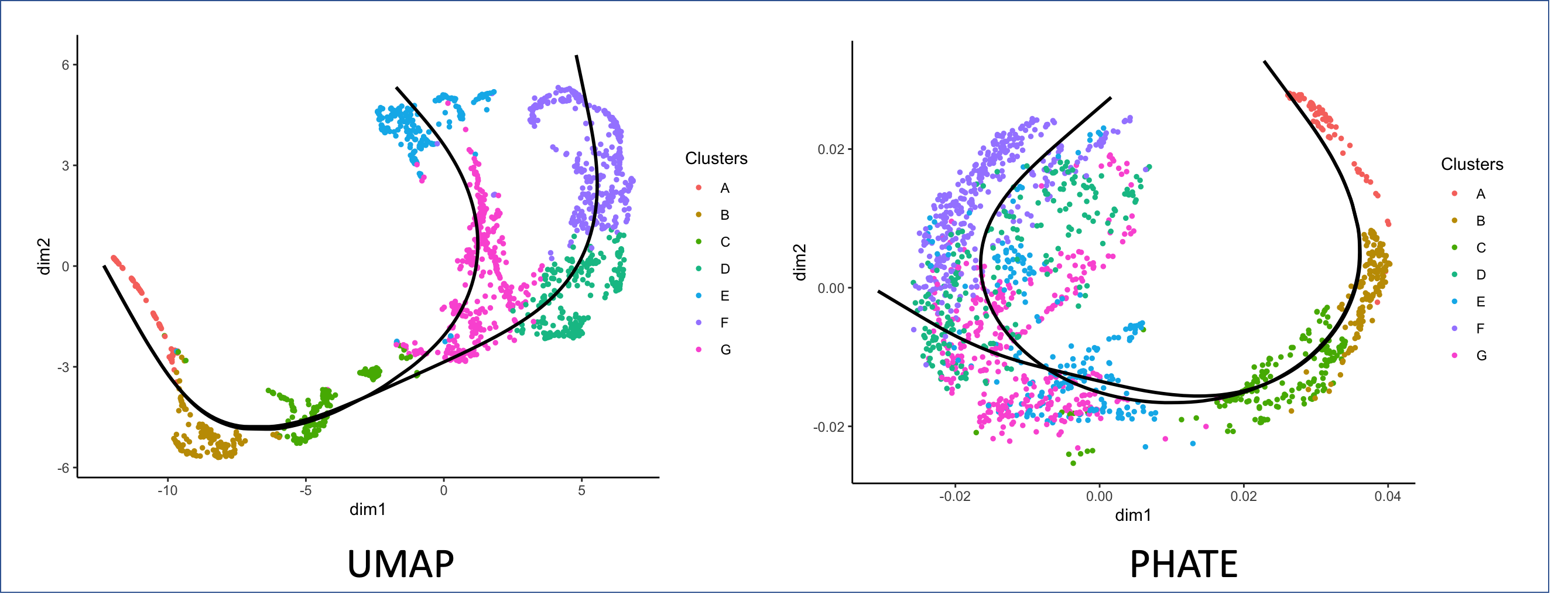
PHATE (Potential of Heat-diffusion for Affinity-based Trajectory Embedding) is a tool for visualizing high dimensional data. PHATE uses a novel conceptual framework for learning and visualizing the manifold to preserve both local and global distances.
To see how PHATE can be applied to datasets such as facial images and single-cell data from human embryonic stem cells, check out our publication in Nature Biotechnology.
PHATE has been implemented in Python >=3.5, MATLAB and R.
- Windows (>= 7), Mac OS X (>= 10.8) or Linux
- Python >= 3.5 or MATLAB (>= 2015a)
All other software dependencies are installed automatically when installing PHATE.
The Python version of PHATE can be installed by running the following from a terminal:
pip install --user phate
Installation of PHATE and all dependencies should take no more than five minutes.
The Python version of PHATE can be installed from GitHub by running the following from a terminal:
git clone --recursive git://github.com/KrishnaswamyLab/PHATE.git
cd PHATE/Python
python setup.py install --user
If you have loaded a data matrix data in Python (cells on rows, genes on columns) you can run PHATE as follows:
import phate
phate_op = phate.PHATE()
data_phate = phate_op.fit_transform(data)
PHATE accepts the following data types: numpy.array, scipy.spmatrix, pandas.DataFrame and anndata.AnnData.
For more information, read the documentation on ReadTheDocs or view our tutorials on GitHub: single-cell RNA-seq, artificial tree. You can also access interactive versions of these tutorials on Google Colaboratory: single-cell RNA-seq, artificial tree.
The MATLAB version of PHATE can be accessed by running the following from a terminal:
git clone --recursive git://github.com/KrishnaswamyLab/PHATE.git
cd PHATE/Matlab
Then, add the PHATE/Matlab directory to your MATLAB path.
Installation of PHATE should take no more than five minutes.
Run any of our run_* scripts to get a feel for PHATE. Documentation is available in the MATLAB help viewer.
In order to use PHATE in R, you must also install the Python package.
If python or pip are not installed, you will need to install them. We recommend Miniconda3 to install Python and pip together, or otherwise you can install pip from https://pip.pypa.io/en/stable/installing/.
First install phate in Python by running the following code from a terminal:
pip install --user phate
Then, install phateR from CRAN by running the following code in R:
install.packages("phateR")
Installation of PHATE and all dependencies should take no more than five minutes.
The development version of PHATE can be installed directly from R with devtools:
if (!suppressWarnings(require(devtools))) install.packages("devtools")
reticulate::py_install("phate", pip=TRUE)
devtools::install_github("KrishnaswamyLab/phateR")
The latest source version of PHATE can be accessed by running the following in a terminal:
git clone --recursive git://github.com/SmitaKrishnaswamy/PHATE.git
cd PHATE/Python
python setup.py install --user
cd ../phateR
R CMD INSTALL
If the phateR folder is empty, you have may forgotten to use the --recursive option for git clone. You can rectify this by running the following in a terminal:
cd PHATE
git submodule init
git submodule update
cd Python
python setup.py install --user
cd ../phateR
R CMD INSTALL
If you have loaded a data matrix data in R (cells on rows, genes on columns) you can run PHATE as follows:
library(phateR)
data_phate <- phate(data)
phateR accepts R matrices, Matrix sparse matrices, data.frames, and any other data type that can be converted to a matrix with the function as.matrix.
For more information and a tutorial, read the phateR README. Documentation is available at https://CRAN.R-project.org/package=phateR/phateR.pdf or in the R help viewer with help(phateR::phate). A tutorial notebook running PHATE on a single-cell RNA-seq dataset is available at http://htmlpreview.github.io/?https://github.com/KrishnaswamyLab/phateR/blob/master/inst/examples/bonemarrow_tutorial.html or in phateR/inst/examples.
If you have any questions or require assistance using PHATE, please contact us at https://krishnaswamylab.org/get-help.































































































































{kind=link}