![]()



Python package for detection of chimerism and contamination in prokaryotic genomes.
- Free software: GNU General Public License v3 or later
- Documentation: GUNC Documentation
Python package for detection of chimerism and contamination in prokaryotic genomes.
License: GNU General Public License v3.0
![]()
Python package for detection of chimerism and contamination in prokaryotic genomes.





































































Hi all,
I have the following error: GUNC gene_counts.json file not found!
Can you help me?
Thanks
For release 1.0.1 the CLI command to download the database does not use a double hyphen, while the installation section documentation states the opposite.
ie. in the docs
$ gunc --download_db /path/to/output/dir/
While the actual command is
$ gunc download_db /path/to/output/dir/
This issue doesn't appear to affect other parts of the documentation.
Hi!
I can only echo the excitement other posters have for using this tool. But I am having trouble getting the conda installation to work for the latest posted versions (1.0.0 and 1.0.1). I do not have admin privileges on the system I am installing to (it is a cluster environment - GNU/Linux x86_64) so following the exact given instruction fails. Here is what I did instead:
This results in the install of v.0.1.2 though.
If I modify step 3 to:
conda install -n gunc -c bioconda gunc=1.0.1
Then I just get a lot of errors:
Collecting package metadata (current_repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: failed with repodata from current_repodata.json, will retry with next repodata source.
Collecting package metadata (repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: |
Found conflicts! Looking for incompatible packages.
This can take several minutes. Press CTRL-C to abort.
failed \
UnsatisfiableError: The following specifications were found to be incompatible with each other:
Output in format: Requested package -> Available versions
Package six conflicts for:
scipy -> mkl-service[version='>=2,<3.0a0'] -> six
numpy -> mkl-service[version='>=2,<3.0a0'] -> six
pandas -> python-dateutil[version='>=2.7.3'] -> six[version='>=1.5']
plotly -> six
plotly -> retrying[version='>=1.3.3'] -> six[version='>=1.7.0']
Package libgcc-ng conflicts for:
pandas -> libgcc-ng[version='>=7.2.0|>=7.3.0']
diamond=2.0.4 -> zlib[version='>=1.2.11,<1.3.0a0'] -> libgcc-ng[version='>=7.2.0|>=7.3.0']
diamond=2.0.4 -> libgcc-ng[version='>=7.5.0']
prodigal -> libgcc-ng[version='>=7.3.0']
numpy -> libgcc-ng[version='>=7.2.0|>=7.3.0']
numpy -> libopenblas[version='>=0.3.2,<0.3.3.0a0'] -> libgcc-ng[version='>=8.2.0']
plotly -> python -> libgcc-ng[version='>=7.2.0|>=7.3.0']
scipy -> libopenblas[version='>=0.3.2,<0.3.3.0a0'] -> libgcc-ng[version='>=8.2.0']
python[version='>=3.6'] -> libgcc-ng[version='>=7.2.0|>=7.3.0']
requests -> python -> libgcc-ng[version='>=7.2.0|>=7.3.0']
scipy -> libgcc-ng[version='>=7.2.0|>=7.3.0']
Package libstdcxx-ng conflicts for:
python[version='>=3.6'] -> libstdcxx-ng[version='>=7.2.0|>=7.3.0']
requests -> python -> libstdcxx-ng[version='>=7.2.0|>=7.3.0']
diamond=2.0.4 -> libstdcxx-ng[version='>=7.5.0']
scipy -> libstdcxx-ng[version='>=7.2.0|>=7.3.0']
numpy -> python[version='>=3.9,<3.10.0a0'] -> libstdcxx-ng[version='>=7.2.0|>=7.3.0']
plotly -> python -> libstdcxx-ng[version='>=7.2.0|>=7.3.0']
pandas -> libstdcxx-ng[version='>=7.2.0|>=7.3.0']
Package _libgcc_mutex conflicts for:
scipy -> libgcc-ng[version='>=7.3.0'] -> _libgcc_mutex=[build=main]
numpy -> libgcc-ng[version='>=7.3.0'] -> _libgcc_mutex=[build=main]
python[version='>=3.6'] -> libgcc-ng[version='>=7.3.0'] -> _libgcc_mutex=[build=main]
diamond=2.0.4 -> libgcc-ng[version='>=7.5.0'] -> _libgcc_mutex=[build=main]
pandas -> libgcc-ng[version='>=7.3.0'] -> _libgcc_mutex=[build=main]
prodigal -> libgcc-ng[version='>=7.3.0'] -> _libgcc_mutex=[build=main]
Package numpy conflicts for:
numpy
pandas -> numpy[version='>=1.11.3,<2.0a0|>=1.12.1,<2.0a0|>=1.13.3,<2.0a0|>=1.14.6,<2.0a0|>=1.15.4,<2.0a0|>=1.16.6,<2.0a0|>=1.9.3,<2.0a0|>=1.9']
scipy -> numpy[version='>=1.11.3,<2.0a0|>=1.14.6,<2.0a0|>=1.16.6,<2.0a0|>=1.15.1,<2.0a0|>=1.9.3,<2.0a0']
Package requests conflicts for:
requests
plotly -> requests
Package pytz conflicts for:
pandas -> pytz[version='>=2017.2']
plotly -> pytz
Package setuptools conflicts for:
plotly -> setuptools
python[version='>=3.6'] -> pip -> setuptools
Package intel-openmp conflicts for:
numpy -> mkl[version='>=2019.4,<2021.0a0'] -> intel-openmp
scipy -> mkl[version='>=2019.4,<2021.0a0'] -> intel-openmp
Any advice would be appreciated!
Thanks,
Craig Herbold
Hallo,
I run gunc on a collection of MAGs and wanted to find out what is the difference between the two dbs progenomes and gtdb. What I saw is first that more MAGs fail when using GTDB. I also checked that more genomes are evaluated at the genus level. Which makes sense as I expect GTDB to have much more genera clusters to evaluate on. But then there are also more genomes evaluated at the Kindom level. Which Doesn't make sense to me?
Do you have any explanation? Is the taxonomic placement more complicated?
What do you generally recommend gtdb or progenomes?
Hello, thank you for creating GUNC! Incredibly helpful for my current project.
Following up on #27 , it seems this has not been resolved. I'm running GUNC v.1.0.5 installed via conda on 4/21/2023, and still get the get_scores.py:114: FutureWarning: iteritems is deprecated and will be removed in a future version. Use .items instead. warning when running gunc run --input_dir my_genomes --threads 28 --out_dir gunc_out, where my_genomes is a directory of fasta files.
Cheers and thanks again!
-Nelson
Hello
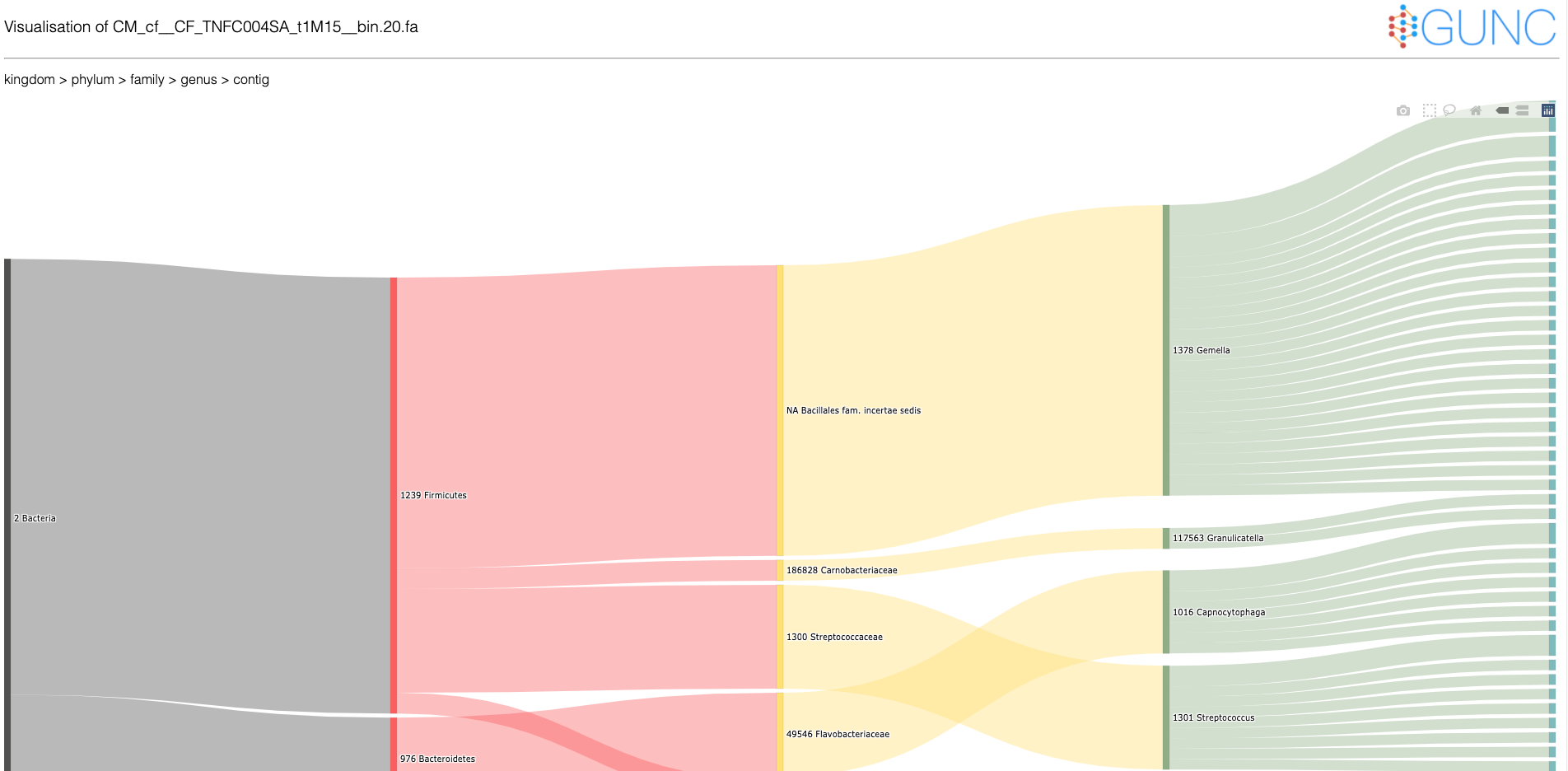
I am confused by the output of gunc - I thought it would be able to identify those contigs which do not match with the rest of the genome - can gunc not do that?
Or at least I thought it would be able to tell me the taxonomic assignments of each contig so I could make the decision myself - does gunc not do this?
It certainly looks from the visualisation (https://grp-bork.embl-community.io/gunc/_images/GUNC_PLOT_example.png) that gunc is able to label contigs - can I get those labels out as a text file?
Thanks
Mick
Dear Gunc Team
I have tried to use GUNC recently to quality control the MAGs that we're building in the lab. For that I also looked into the internal diamond command for which you state the -k 1 will be retaining best hits. However setting -k 1 will search for the best hits but report only one. Setting --top 0 on the other hand will report all best hits. I'm not sure about the internals of GUNC but this could potentially inflate false positive detection of chimeras?
Example:
diamond .... --max-target-seqs 1 --> will find the best hit but ignore 2 other hits that score equally well
GENE GB_GCA_001915545.1 100.0 221 0 0 1 221 1 221 1.4e-122 447.6
diamond .... --top 0 --> will find multiple best hits
GENE GB_GCA_001915545.1 100.0 221 0 0 1 221 1 221 1.4e-122 447.6
GENE GB_GCA_900555085.1 100.0 221 0 0 1 221 1 221 1.4e-122 447.6
GENE RS_GCF_000012825.1 100.0 221 0 0 1 221 1 221 1.4e-122 447.6
Hello,
I see the statistics on GTDB, the amount of genomes doubled in the database since release 95, which is used in gunc. Is there a plan to update the GTDB database?
Best,
Pavlo
Hi,
I have install gunc with conda.
When I run gunc with the command :
gunc run -i /beegfs/project/nega/assembly/re-run-article/hifiasm_pacbio/hifiasm/assembly_coiffaiti_hifiasm.asm.bp.p_ctg.fa -r /beegfs/data/gdebaecker/soft/gunc_db/gunc_db_progenomes2.1.dmnd -t 16
I got the following error :
'''
Illegal instruction
Traceback (most recent call last):
File "/beegfs/data/gdebaecker/myconda/conda-env/gunc/bin/gunc", line 10, in
sys.exit(main())
^^^^^^
File "/beegfs/data/gdebaecker/myconda/conda-env/gunc/lib/python3.11/site-packages/gunc/gunc.py", line 712, in main
start_checks()
File "/beegfs/data/gdebaecker/myconda/conda-env/gunc/lib/python3.11/site-packages/gunc/gunc.py", line 228, in start_checks
diamond_ver = external_tools.check_diamond_version()
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File "/beegfs/data/gdebaecker/myconda/conda-env/gunc/lib/python3.11/site-packages/gunc/external_tools.py", line 169, in check_diamond_version
subprocess.check_output(
File "/beegfs/data/gdebaecker/myconda/conda-env/gunc/lib/python3.11/subprocess.py", line 466, in check_output
return run(*popenargs, stdout=PIPE, timeout=timeout, check=True,
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File "/beegfs/data/gdebaecker/myconda/conda-env/gunc/lib/python3.11/subprocess.py", line 571, in run
raise CalledProcessError(retcode, process.args,
subprocess.CalledProcessError: Command 'diamond --version' returned non-zero exit status 132.
''''
but when I try the command : diamond --version --> diamond version 2.0.4
Do you know how I can correct this ?
Thanks in advance,
Gautier
FileNotFoundError: [Errno 2] No such file or directory: '/conda/9ff217aa/lib/python3.9/site-packages/gunc/data/genome2taxonomy_ref.tsv'
I installed diamond prodigal and gunc using conda and get this error. I would really appreciate your thoughts.
Hi
While running prok-quality I had encountered possible false positives for genomes as "chimeric" according to support metashot/prok-quality#5. They're all Vibrio cholerae genomes downloaded from NCBI.
genome_info.zip
Thanks for the great tool! Minor issue - when running gunc merge_checkm, it appears that the checkm output file needs to contain specific columns that are only created when checkm qa is run with the -o 2 (extended summary of bin quality) option. Attempting to run the merge command on the default checkm output gives the following error:
Traceback (most recent call last):
File "/labs/asbhatt/bsiranos/miniconda3/bin/gunc", line 8, in <module>
sys.exit(main())
File "/labs/asbhatt/bsiranos/miniconda3/lib/python3.7/site-packages/gunc/gunc.py", line 529, in main
merge_checkm(args)
File "/labs/asbhatt/bsiranos/miniconda3/lib/python3.7/site-packages/gunc/gunc.py", line 510, in merge_checkm
merged = checkm_merge.merge_checkm_gunc(args.checkm_file, args.gunc_file)
File "/labs/asbhatt/bsiranos/miniconda3/lib/python3.7/site-packages/gunc/checkm_merge.py", line 60, in merge_checkm_gunc
'checkM.genome_size': checkmdata['Genome size (bp)'],
KeyError: 'Genome size (bp)'
A specificaion in the docs, or compatability with the defaul checkm output is all that's required.
Hi!
Thank you for the very cool and useful tool!
I would like to know if it is possible to have access to the synthetic data you generated to benchmark GUNC on the different chimerism scenarios.
Thanks again!
Chiara
Dear @fullama ,
I get the error
gunc download_db ./ usage: gunc [-h] -d (-i | -g ) [-p] [-t] [-o] [-s] [-v] gunc: error: the following arguments are required: -d/--database_file
In both cases, running
gunc download_db ./
or
gunc --download_db ./
What am I doing wrong?
Bests,
Ulrike
the default setting of gunc is use phylum level as maxCSS. but i find that the phylum level is faulse in pass.GUNC, but the phylum level is true in pass.GUNC, so which result is more credible?
Hello,
I met the panda key error (raise KeyError(key) from err KeyError: 'contig') during the diamond step, and gunc just quit running. In the "diamond_output" folder, it produced two outputs for each query fasta file, XX.diamond.progenomes_2.1.out (not empty) and XX..diamond.out (empty). There were also a few unreadable files with names like ".nfs000000000447d97c0001a098" within "diamond_output".
Could you please let me know what went wrong?
Thank you
Hello
I ran GUNC v1.0.5 on some bins and all of the values in the result tsv file were NAN since there is no genes can be mapped by diamond. I wonder if there exist some bugs in it?
<style> </style>| genome | n_genes_called | n_genes_mapped | n_contigs | taxonomic_level | proportion_genes_retained_in_major_clades | genes_retained_index | clade_separation_score | contamination_portion | n_effective_surplus_clades | mean_hit_identity | reference_representation_score | pass.GUNC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 3235 | 0 | 0 | kingdom | nan | nan | nan | nan | nan | nan | nan | nan |
| 1 | 1075 | 0 | 0 | kingdom | nan | nan | nan | nan | nan | nan | nan | nan |
| 10 | 5890 | 0 | 0 | kingdom | nan | nan | nan | nan | nan | nan | nan | nan |
| 100 | 12397 | 0 | 0 | kingdom | nan | nan | nan | nan | nan | nan | nan | nan |
| 101 | 5185 | 0 | 0 | kingdom | nan | nan | nan | nan | nan | nan | nan | nan |
conda install no longer finds diamond=2.0.4
Do you know how I could download that? (Also working in an HPCC where I am not the administrator.)
Originally posted by @ggavelis in #5 (comment)
We are using conda 4.7.5 and when installing v1.0.3 of GUNC, the
(gunc)$ gunc download_db ./
[START] 09:36:08 2021-07-16
[INFO] DB downloading...
Traceback (most recent call last):
File "/home/ecoli/anaconda/envs/gunc/bin/gunc", line 10, in <module>
sys.exit(main())
File "/home/ecoli/anaconda/envs/gunc/lib/python3.6/site-packages/gunc/gunc.py", line 561, in main
gunc_database.get_db(args.path, args.database)
File "/home/ecoli/anaconda/envs/gunc/lib/python3.6/site-packages/gunc/gunc_database.py", line 108, in get_db
download_file(gz_file_url, gz_file_path)
File "/home/ecoli/anaconda/envs/gunc/lib/python3.6/site-packages/gunc/gunc_database.py", line 38, in download_file
with requests.get(file_url, stream=True) as r:
AttributeError: __enter__
Seeing other peoples problems I tried downgrading to version 1.0.1, but got the same error;
(gunc)$ gunc download_db ./
[START] 09:39:26 2021-07-16
[INFO] DB downloading...
Traceback (most recent call last):
File "/home/ecoli/anaconda/envs/gunc/bin/gunc", line 10, in <module>
sys.exit(main())
File "/home/ecoli/anaconda/envs/gunc/lib/python3.6/site-packages/gunc/gunc.py", line 520, in main
gunc_database.get_db(args.path)
File "/home/ecoli/anaconda/envs/gunc/lib/python3.6/site-packages/gunc/gunc_database.py", line 104, in get_db
download_file(gz_file_url, gz_file_path)
File "/home/ecoli/anaconda/envs/gunc/lib/python3.6/site-packages/gunc/gunc_database.py", line 38, in download_file
with requests.get(file_url, stream=True) as r:
AttributeError: __enter__
Is there any solution to this?
I keep getting this error "Failed to run Diamond gunc_out/gene_calls/merged.genecalls.faa"
gunc run -r gunc_db_progenomes2.1.dmnd -i genome4.fna -o gunc_out -e .fna
When I check the gene_calls folder, I find genome4.genecalls.faa. Is this the issue that the program cannot find merged.....?
GUNC is a useful software, but the database version of it seems too old. Is there any plan to update GUNC database? Or How to make a database by users themselves? I found that some one have also asked the question:
#40 (comment)
Minor issue:
Unhelpful error when trying to download db into non-existing directory.
Explanation:
There is no consistency amongst bioinformatics software whether a target directory has to or is not allowed to exist prior to running the command. In the case of "gunc download_db" the target dir needs to be created in advance, but the error isn't particularly helpful.
version:
version 1.0.0 through bioconda on a linux system
Command run
gunc download_db /export/data1/db/GUNC
Result:
[START] 15:09:44 2020-12-17
[INFO] DB downloading...
Traceback (most recent call last):
File "/export/data1/sw/anaconda3-2019.07/envs/dRep/bin/gunc", line 10, in
sys.exit(main())
File "/export/data1/sw/anaconda3-2019.07/envs/dRep/lib/python3.6/site-packages/gunc/gunc.py", line 507, in main
gunc_database.get_db(args.path)
File "/export/data1/sw/anaconda3-2019.07/envs/dRep/lib/python3.6/site-packages/gunc/gunc_database.py", line 101, in get_db
download_file(gz_file_url, gz_file_path)
File "/export/data1/sw/anaconda3-2019.07/envs/dRep/lib/python3.6/site-packages/gunc/gunc_database.py", line 39, in download_file
with open(out_file, 'wb') as f:
FileNotFoundError: [Errno 2] No such file or directory: '/export/data1/db/GUNC/gunc_db_2.0.4.dmnd.gz'
Hi,
I just downloaded gunc using a conda install (v 1.0.4) and and diamond keeps failing because it is looking for "merged.genecalls.faa" and prodigal has created called genes file "input_genome_bin.faa". my command I've tried is: gunc run -i input_genome_bin.fa -r path/to/database/gunc_db_progenomes2.1.dmnd -t 2.
Seems like this is hard coded in and not something we can specify? But I might be missing something.
Thanks in advance.
I'm unable to install gunc in a separate conda environment for gunc on Ubuntu 16.04. The commands I tried and their outputs on the terminal are as below:
Command1 conda install -c bioconda gunc
Output Collecting package metadata (current_repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: failed with repodata from current_repodata.json, will retry with next repodata source.
Collecting package metadata (repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: -
Found conflicts! Looking for incompatible packages.
This can take several minutes. Press CTRL-C to abort.
failed
UnsatisfiableError:
Command2 conda install -c bioconda/label/broken gunc
Output Collecting package metadata (current_repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: failed with repodata from current_repodata.json, will retry with next repodata source.
Collecting package metadata (repodata.json): done
Solving environment: failed with initial frozen solve. Retrying with flexible solve.
Solving environment: \
Found conflicts! Looking for incompatible packages.
This can take several minutes. Press CTRL-C to abort.
failed
ResolvePackageNotFound:
Please let me know what should I do?
Many thanks
Amit
Hi,
I have a question regarding the threshold CSS value and its relationship with genomes RRS and their inclusion in GUNC database.
I'm checking the quality of a set of genomes and in some cases I obtain CSS values between 0.48 and 0.50 which given the default CSS threshold are being flagged as contaminated.
In some cases these genomes present RRS values above 0.5 (in some cases up to 0.97). In fact, some of these genomes have been downloaded from RefSeq while others have a GTDB-Tk classification included in the reference database that I'm using (ProGenomes). Here is the table with the results:
| CheckM2 completeness | CheckM2 contamination | CSS | GUNC contamination | GUNC effective surplus clades | GUNC mean hit identity | RRS | |
|---|---|---|---|---|---|---|---|
| GCA_947444635.1 | 60.28 | 0.08 | 0.07 | 0.05 | 0.1 | 0.72 | 0.6 |
| GCA_947470005.1 | 75.45 | 0.15 | 0.27 | 0.07 | 0.16 | 0.67 | 0.55 |
| GCA_027437095.1 | 88.44 | 2.67 | 0.33 | 0.03 | 0.06 | 0.69 | 0.54 |
| GCF_001467945.1 | 99.94 | 0.4 | 0.44 | 0.05 | 0.11 | 0.99 | 0.97 |
| GCF_900461585.1 | 99.96 | 0.4 | 0.44 | 0.05 | 0.11 | 0.99 | 0.97 |
| GCF_900639925.1 | 99.96 | 0.37 | 0.47 | 0.05 | 0.11 | 0.99 | 0.97 |
| GCA_903907265.1 | 52.32 | 0.03 | 0.47 | 0.05 | 0.11 | 0.62 | 0.52 |
| GCA_003507515.1 | 76.3 | 0.89 | 0.48 | 0.03 | 0.05 | 0.68 | 0.58 |
| GCF_001736145.1 | 99.93 | 1.78 | 0.48 | 0.05 | 0.11 | 0.99 | 0.97 |
| GCA_002352055.1 | 99.95 | 4.17 | 0.52 | 0.05 | 0.1 | 0.72 | 0.64 |
| GCA_027358185.1 | 94.09 | 1.29 | 0.59 | 0.03 | 0.06 | 0.67 | 0.57 |
| GCF_001468135.1 | 100 | 2.54 | 0.6 | 0.03 | 0.07 | 0.99 | 0.96 |
| GCA_903885895.1 | 94.68 | 5.11 | 0.63 | 0.1 | 0.23 | 0.61 | 0.44 |
| GCA_945865355.1 | 86.73 | 3.6 | 0.67 | 0.02 | 0.05 | 0.69 | 0.61 |
| GCF_900452545.1 | 100 | 1.29 | 0.7 | 0.04 | 0.09 | 0.99 | 0.97 |
| GCF_900639855.1 | 100 | 1.28 | 0.7 | 0.04 | 0.09 | 0.99 | 0.97 |
| GCA_947474165.1 | 97.55 | 0.22 | 0.72 | 0.04 | 0.08 | 0.67 | 0.56 |
| GCF_001467695.1 | 100 | 1.29 | 0.73 | 0.04 | 0.09 | 0.99 | 0.97 |
| GCF_900639975.1 | 100 | 3.29 | 0.74 | 0.03 | 0.07 | 0.99 | 0.96 |
| GCA_903842685.1 | 73.45 | 4.49 | 0.82 | 0.12 | 0.28 | 0.63 | 0.45 |
| GCA_947485955.1 | 91.54 | 4.96 | 0.86 | 0.03 | 0.05 | 0.67 | 0.52 |
| GCA_903901775.1 | 67.23 | 1.56 | 1 | 0.04 | 0.08 | 0.69 | 0.53 |
From what I understand in Figure S12 of the manuscript, the optimal CSS threshold for genomes which are included within the reference database is slightly higher than the for out-of-reference genomes (with a peak around 0.475) and for this reason I was wondering if it would make sense to not discard the genomes genomes with CSS between 0.45 and 0.48-0.50.
Thanks for the support and the great tool!
Hi,
for some samples I had this error:
...
15:45:52 : No genes mapped to reference: SemiBin_95
15:45:52 : No genes mapped to reference: SemiBin_96
15:45:52 : No genes mapped to reference: SemiBin_97
15:45:52 : No genes mapped to reference: SemiBin_98
15:45:52 : No genes mapped to reference: SemiBin_99
15:45:52 : No genes mapped to reference: SemiBin_9
15:45:52 : No diamond output files.
When I list my diamond output files:
SemiBin_101.diamond.out
SemiBin_100.diamond.out
SemiBin_1005.diamond.out
SemiBin_1004.diamond.out
SemiBin_0.diamond.out
...
SemiBin_101.diamond.progenomes_2.1.out
SemiBin_100.diamond.progenomes_2.1.out
SemiBin_1005.diamond.progenomes_2.1.out
SemiBin_1004.diamond.progenomes_2.1.out
SemiBin_0.diamond.progenomes_2.1.out
I checked the source code, and the issue looks like to be triggered here:
Line 732 in 0e64c39
that I guess should be changed (in my case) to:
diamond_outdir, "diamond_output", f"{basename}.diamond.progenomes_2.1.out"
that is only triggered when there is one diamond output file missing
Line 693 in 0e64c39
I don't know if this is a bug. Maybe the database name changed, modifying the output name? Not sure.
Hi,
I am using gunc v1.0.2 in a fresh Conda environment to perform chimerism checks on a few test genomes. I have already generated Prodigal calls, so I am providing them as the input fasta after setting the gene_calls flag.
Initial command:
gunc run --db_file /home/annotator/database/gunc_db_gtdb95.dmnd --input_fasta proteins.faa --file_suffix .faa --gene_calls --threads 64 --out_dir /tmp/tmppbxk2bwz
After DIAMOND finishes running, I consistently get the following error:
Traceback (most recent call last):
File "/opt/conda/envs/gunc_env/bin/gunc", line 10, in <module>
sys.exit(main())
File "/opt/conda/envs/gunc_env/lib/python3.9/site-packages/gunc/gunc.py", line 567, in main
run(args)
File "/opt/conda/envs/gunc_env/lib/python3.9/site-packages/gunc/gunc.py", line 475, in run
gunc_output = run_gunc(diamond_outfiles, genes_called, args.out_dir,
File "/opt/conda/envs/gunc_env/lib/python3.9/site-packages/gunc/gunc.py", line 389, in run_gunc
gene_call_count = genes_called[basename]
KeyError: 'proteins.faa'
Since the basename is generated by this line of code: basename = os.path.basename(diamond_file).split('.diamond.')[0] I am not sure of the exact source of the error.
Hi all,
I dont know if this is the correct place to make this question but.. I would like to know the way to run GUNC with a large dataset of MAGs (700).
I tried with:
for i in cat list.txt; do gunc run -t 20 -i $i -r db_gunc/gunc_db_progenomes2.1.dmnd -o gunc/; done
The problem is that the TSV table (with the clade_separation_score) is overwritten after each round and I need to keep the previous one as well
Thank you very much!
Pat
Hello,
Thanks for developing the tool. I have some questions and/or suggestions:
[START] 11:41:09 2020-12-18
Traceback (most recent call last):
File "/home/aloliveira/anaconda3/envs/gunc/bin/gunc", line 10, in <module>
sys.exit(main())
File "/home/aloliveira/anaconda3/envs/gunc/lib/python3.9/site-packages/gunc/gunc.py", line 512, in main
run(args)
File "/home/aloliveira/anaconda3/envs/gunc/lib/python3.9/site-packages/gunc/gunc.py", line 403, in run
check_for_duplicate_filenames(fnas, args.file_suffix)
UnboundLocalError: local variable 'fnas' referenced before assignment
The command line that I've used: gunc run -g Gene_predictions.faa -r gunc_db_2.0.4.dmnd
Swapped fields concerning the --input_dir and --input_file parameters
GUNC RUN
Run chimerism detection.
Required Flags
--db_file Path to the GUNC database file. Can be set as environment variable GUNC_DB.
One of the following is required. If contigs (fna) are supplied the gene calls will be done using prodigal with option “-p meta”.
**--input_dir Input file in FASTA fna format.**
--file_suffix Only needed if suffix of files in --input_dir is not the default .fa.
**--input_file Input file contining paths to FASTA fna format files.**
--input_fna Input file in FASTA fna format.
--gene_calls Input genecalls FASTA faa format.
--use_species_level Allow species level to be picked as maxCSS. Default: False
--min_mapped_genes Dont calculate GUNC score if number of mapped genes is below this value. Default: 11
Thanks again for this really useful tool,
André
Dear Gunc Team,
I am using GUNC v1.0.5, and want to ask a question about the --file_suffix. The suffix of my input files is .fna, and some genomes from NCBI may contain .fna in the middle of genome names. If providing with --input_dir and --file_suffix .fna, I am wondering whether GUNC could make right action on those kind of genomes that contains .fna in the middle of names? So I provide the --input_file with the path of each genome, may I ask whether --file_suffix .fna is still needed when --input_file is provide? Or any other suggestions?
Many thanks
Wang
Hi,
using the pip installation inside a new conda env and version 1.0.1, I got this error while running the command : gunc download_db ./
[START] 19:17:04 2021-01-06
[INFO] DB downloading...
Traceback (most recent call last):
File "/home/michoug/miniconda3/envs/gunc/bin/gunc", line 10, in <module>
sys.exit(main())
File "/home/michoug/miniconda3/envs/gunc/lib/python3.6/site-packages/gunc/gunc.py", line 520, in main
gunc_database.get_db(args.path)
File "/home/michoug/miniconda3/envs/gunc/lib/python3.6/site-packages/gunc/gunc_database.py", line 104, in get_db
download_file(gz_file_url, gz_file_path)
File "/home/michoug/miniconda3/envs/gunc/lib/python3.6/site-packages/gunc/gunc_database.py", line 38, in download_file
with requests.get(file_url, stream=True) as r:
AttributeError: __enter_
run_gunc exits due to the lack of a series.iteritems method.
the default setting of gunc is use phylum level as maxCSS. but i find that the phylum level is faulse in pass.GUNC, but the phylum level is true in pass.GUNC of same bin, so which result is more credible?
Hello,
I was in the process of creating a nextflow module for GUNC for the nf-core pipeline initiative.
Each nf-core module offers both the use of conda but also biocontainers (docker or singularity, among others), to improve portability and reproducibility.
Unfortunately, during testing of the gunc run nexflow module, while the test ran with conda, it failed for both docker and singularity, with the following error:
Command error:
[ERROR] zgrep not found..
After investigating, it appears to be that the base (extremely minimal) image that biocontainers use do not include zgrep.
I initially looked to see if zgrep existed as a conda-recipe to add it the GUNC recipe as a dependency, but wierdly (and sadly) this does not seem to be the case.
I was wondering if there was a specific reason why zgrep is used, or whether it could be replaced with a gzip -c <> | grep like system, to get around this?
The alternative would be a lot of upstream work (getting zgrep into a conda recipe, and/or then requesting the base biocontainer image to be updated), both of which something which is out of my expertise nor authority).
Cheers,
James
it seems that my computer don't support diamond 20.0..4, error info:
terminate called after throwing an instance of 'std::runtime_error'
what(): CPU does not support SSSE3. Please compile the software from source.
Aborted (core dumped)
Hi. I'm wondering if there are plans to extend GUNC to report contigs that are likely potential contamination based on having a substantial number of incongruent genes?
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.
{kind=link}