



- Introduction
- Installation
- Testing
- Inputs
- Outputs
- EDTA usage
- panEDTA usage
- Benchmark
- Citations
- Other resources
- Questions and Issues
- Acknowledgements
This package is developed for automated whole-genome de-novo TE annotation and benchmarking the annotation performance of TE libraries.
The EDTA package was designed to filter out false discoveries in raw TE candidates and generate a high-quality non-redundant TE library for whole-genome TE annotations. Selection of initial search programs were based on benckmarkings on the annotation performance using a manually curated TE library in the rice genome.
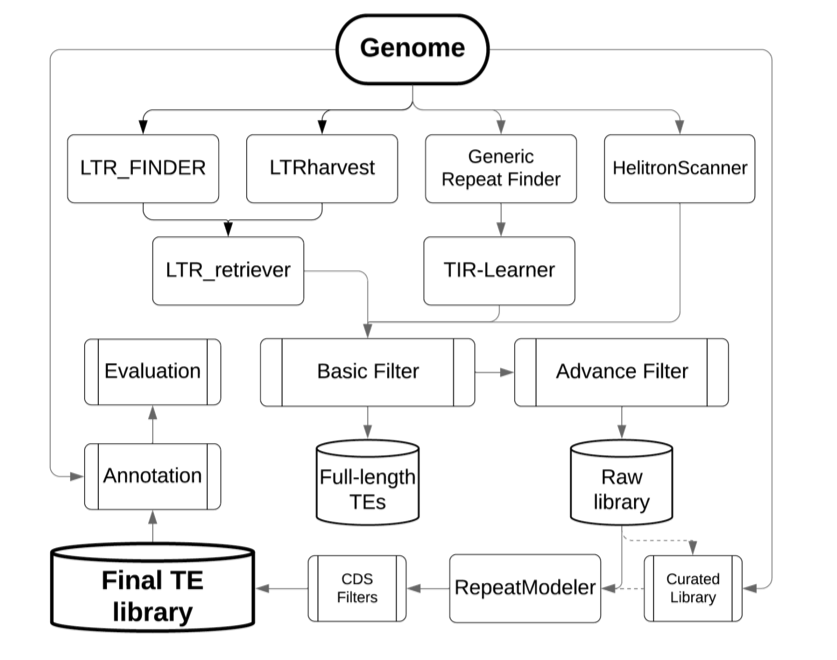
To benchmark the annotation quality of a new library/method, I have provided the TE annotation with the curated rice TE library (v7.0.0) for the rice genome (TIGR7/MSU7 version). You may use the lib-test.pl script to compare the annotation performance of your method/library to the methods we have tested (usage shown below).
For pan-genome annotations, you need to annotate each genome with EDTA, generate a pan-genome library, then reannotate each genome with the pan-genome library. Please refer to this example for details. A sequential version of panEDTA is also included in this package.
There are several ways to install EDTA. You just need to find the one that works for your system. If you are not using macOS, you may try the conda approach before the Singularity approach.
Install with conda/mamba (Linux64)
Recommend to ceate a dedicated environment for EDTA:
conda create -n EDTA
conda activate EDTA
mamba install -c conda-forge -c bioconda edta
Other ways to install with conda/mamba...
- Install with the yml file:
Download the latest EDTA:
git clone https://github.com/oushujun/EDTA.git
Find the yml file in the EDTA folder and run:
mamba env create -f EDTA_2.2.x.yml
The default conda env name is EDTA2 specified by the first line of the yml file. You may change that to different names.
- Install by specifying all dependencies:
mamba create -n EDTA2.2 -c conda-forge -c bioconda -c r annosine2 biopython blast cd-hit coreutils genericrepeatfinder genometools-genometools glob2 h5py==3.9 keras==2.11 ltr_finder ltr_retriever mdust multiprocess muscle openjdk pandas perl perl-text-soundex pyarrow python r-base r-dplyr regex repeatmodeler r-ggplot2 r-here r-tidyr scikit-learn swifter tensorflow==2.11 tesorter
Usage:
conda activate EDTA
perl EDTA.pl
You can use the conda ENV to execute the latest EDTA from GitHub:
git clone https://github.com/oushujun/EDTA.git
perl ./EDTA/EDTA/pl
Install with Singularity (good for HPC users)
SINGULARITY_CACHEDIR=./
export SINGULARITY_CACHEDIR
`singularity pull EDTA.sif docker://quay.io/biocontainers/edta:<tag>`
Visit BioContainers repository for a list of available tags (e.g., 2.2.0--hdfd78af_1).
Usage:
export PYTHONNOUSERSITE=1
singularity exec {path}/EDTA.sif EDTA.pl --genome genome.fa [other parameters]
Where {path} is the path you build the EDTA singularity image.
Install with Docker (good for root/macOS/Apple M-chip users)
sudo docker pull quay.io/biocontainers/edta:<tag>
Usage:
sudo docker run -v $PWD:/in -w /in quay.io/biocontainers/edta:<tag> EDTA.pl --genome genome.fa [other parameters]
Visit BioContainers repository for a list of available tags (e.g., 2.2.0--hdfd78af_1).
Note: Because only the current directory is mounted to the EDTA docker container, you have to copy all needed files to the current directory and provide them to EDTA without path specifications. Even providing the absolute path to the file located in this folder won't work. Softlinked files are considered "with path" and won't work. Similarily, specifying your own versions of dependency programs (i.e., repeatmasker, repeatmodeler) won't work because they have paths.
You should test the EDTA pipeline with a 1-Mb toy genome, which takes about five mins. If your test finishs without any errors (warnings are OK), then EDTA should be correctly installed. If the test is OK but you encounter errors with your data, you should check your own data for any formating/naming mistakes.
cd ./EDTA/test
perl ../EDTA.pl --genome genome.fa --cds genome.cds.fa --curatedlib ../database/rice7.0.0.liban --exclude genome.exclude.bed --overwrite 1 --sensitive 1 --anno 1 --threads 10
If your test fails, you may check out this collection of issues for possible reasons and solutions. If none works, you may open a new issue.
Required: The genome file [FASTA]. Please make sure sequence names are short (<=13 characters) and simple (i.e, letters, numbers, and underscore).
Optional:
- Coding sequence of the species or a closely related species [FASTA]. This file helps to purge gene sequences in the TE library.
- Known gene positions of this version of the genome assembly [BED]. Coordinates specified in this file will be excluded from TE annotation to avoid over-masking.
- Curated TE library of the species [FASTA]. This file is trusted 100%. Please make sure it's curated. If you only have a couple of curated sequences, that's also good. It doesn't need to be complete. Providing curated TE sequences, especially for those under-annotated TE types (i.e., SINEs and LINEs), will greatly improve the annotation quality. For more information, please visit this wiki page: How to prepare a curated library to maximize the efficacy of EDTA
A non-redundant TE library: $genome.mod.EDTA.TElib.fa. The curated library will be included in this file if provided. The rice library will be (partially) included if --force 1 is specified. TEs are classified into the superfamily level and using the three-letter naming system reported in Wicker et al. (2007). Each sequence can be considered as a TE family. To convert between classification systems, please refer to the TE sequence ontology file.
Optional 1:
- Novel TE families: $genome.mod.EDTA.TElib.novel.fa. This file contains TE sequences that are not included in the curated library (
--curatedlibrequired).
Optional 2, when you specify the --anno 1 parameter, you will get:
2. Whole-genome TE annotation: $genome.mod.EDTA.TEanno.gff3. This file contains both structurally intact and fragmented TE annotations.
3. Summary of whole-genome TE annotation: $genome.mod.EDTA.TEanno.sum.
4. Low-threshold TE masking: $genome.mod.MAKER.masked. This is a genome file with only long TEs (>=1 kb) being masked. You may use this for de novo gene annotations. In practice, this approach will reduce overmasking for genic regions, which can improve gene prediction quality. However, initial gene models should contain TEs and need further filtering.
5. Annotation inconsistency for simple TEs: $genome.mod.EDTA.TE.fa.stat.redun.sum.
6. Annotation inconsistency for nested TEs: $genome.mod.EDTA.TE.fa.stat.nested.sum.
7. Oveall annotation inconsistency: $genome.mod.EDTA.TE.fa.stat.all.sum.
You got a genome and you want to get a high-quality TE annotation:
perl EDTA.pl [options]
--genome [File] The genome FASTA file. Required.
--species [Rice|Maize|others] Specify the species for identification of TIR candidates. Default: others
--step [all|filter|final|anno] Specify which steps you want to run EDTA.
all: run the entire pipeline (default)
filter: start from raw TEs to the end.
final: start from filtered TEs to finalizing the run.
anno: perform whole-genome annotation/analysis after TE library construction.
--overwrite [0|1] If previous results are found, decide to overwrite (1, rerun) or not (0, default).
--cds [File] Provide a FASTA file containing the coding sequence (no introns, UTRs, nor TEs) of this genome or its close relative.
--curatedlib [file] Provided a curated library to keep consistant naming and classification for known TEs.
All TEs in this file will be trusted 100%, so please ONLY provide MANUALLY CURATED ones here.
This option is not mandatory. It's totally OK if no file is provided (default).
--sensitive [0|1] Use RepeatModeler to identify remaining TEs (1) or not (0, default).
This step is very slow and MAY help to recover some TEs.
--anno [0|1] Perform (1) or not perform (0, default) whole-genome TE annotation after TE library construction.
--rmout [File] Provide your own homology-based TE annotation instead of using the EDTA library for masking.
File is in RepeatMasker .out format. This file will be merged with the structural-based TE annotation. (--anno 1 required).
Default: use the EDTA library for annotation.
--evaluate [0|1] Evaluate (1) classification consistency of the TE annotation. (--anno 1 required). Default: 0.
This step is slow and does not affect the annotation result.
--exclude [File] Exclude regions (bed format) from TE masking in the MAKER.masked output. Default: undef. (--anno 1 required).
--u [float] Neutral mutation rate to calculate the age of intact LTR elements.
Intact LTR age is found in this file: *EDTA_raw/LTR/*.pass.list. Default: 1.3e-8 (per bp per year, from rice).
--threads|-t [int] Number of theads to run this script (default: 4)
--help|-h Display this help info
Identify intact elements of a paticular TE type:
1.Get raw TEs from a genome (specify -type ltr|tir|helitron in different runs)
perl EDTA_raw.pl [options]
--genome [File] The genome FASTA
--species [Rice|Maize|others] Specify the species for identification of TIR candidates. Default: others
--type [ltr|tir|helitron|line|sine|all]
Specify which type of raw TE candidates you want to get. Default: all
--overwrite [0|1] If previous results are found, decide to overwrite (1, rerun) or not (0, default).
--threads|-t [int] Number of theads to run this script
--help|-h Display this help info
2.Finish the rest of the EDTA analysis (specify -overwrite 0 and it will automatically pick up existing results in the work folder)
perl EDTA.pl --overwrite 0 [options]
- It's never said enough. You should tidy up all your sequence names before ANY analysis. Keep them short, simple, and unique.
- Run it in a fast drive (i.e., SSD) because RepeatMasker/RepeatModeler is I/O intense.
- Check out the Wiki page for more information and frequently asked questions.
This is the serial version of panEDTA. Each genome will be annotated sequentially and then combined with the panEDTA functionality. Existing EDTA annotation of genomes (EDTA run with --anno 1) will be recognized and reused. A way to acclerate the pan-genome annotation is to execute EDTA annotation of each genomes separately and in parallel, then execute panEDTA to finish the remaining of the runs. You may want to save the GFF files and the sum file of the EDTA results of each genome because they will be overwritten by panEDTA. To help filtering out gene-related sequences, at least one CDS file is required. Please read wiki for the CDS requirement. You may want to check out the toy example in the ./test folder to get familiarized.
sh panEDTA.sh -g genome_list.txt -c cds.fasta -t 10
-g A list of genome files with paths accessible from the working directory.
Required: You can provide only a list of genomes in this file (one column, one genome each row).
Optional: You can also provide both genomes and CDS files in this file (two columns, one genome and one CDS each row).
Missing of CDS files (eg, for some or all genomes) is allowed.
-c Required. Coding sequence files in fasta format.
The CDS file provided via this parameter will fill in the missing CDS files in the genome list.
If no CDS files are provided in the genome list, then this CDS file will be used on all genomes.
-l Optional. A manually curated, non-redundant library following the RepeatMasker naming format.
-f Minimum number of full-length TE copies in individual genomes to be kept as candidate TEs for the pangenome.
Lower is more inclusive, and will ↑ library size, ↑ sensitivity, and ↑ inconsistency.
Higher is more stringent, and will ↓ library size, ↓ sensitivity, and ↓ inconsistency.
Default: 3.
-t Number of CPUs to run panEDTA. Default: 10.
If you developed a new TE method/got a TE library and want to compare it's annotation performance to the methods we have tested, you can:
1.annotate the rice genome with your test library:
RepeatMasker -e ncbi -pa 36 -q -no_is -norna -nolow -div 40 -lib custom.TE.lib.fasta -cutoff 225 rice_genome.fasta
2.Test the annotation performance of a particular TE category.
perl lib-test.pl -genome genome.fasta -std genome.stdlib.RM.out -tst genome.testlib.RM.out -cat [options]
-genome [file] FASTA format genome sequence
-std [file] RepeatMasker .out file of the standard library
-tst [file] RepeatMasker .out file of the test library
-cat [string] Testing TE category. Use one of LTR|nonLTR|LINE|SINE|TIR|MITE|Helitron|Total|Classified
-N [0|1] Include Ns in total length of the genome. Defaule: 0 (not include Ns).
-unknown [0|1] Include unknown annotations to the testing category. This should be used when
the test library has no classification and you assume they all belong to the
target category specified by -cat. Default: 0 (not include unknowns)
eg.
perl lib-test.pl -genome rice_genome.fasta -std ./EDTA/database/Rice_MSU7.fasta.std7.0.0.out -tst rice_genome.fasta.test.out -cat LTR
Note: the -std and -tst files should be named differently even they are placed in different folders.
Please cite our paper if you find EDTA useful:
Ou S., Su W., Liao Y., Chougule K., Agda J. R. A., Hellinga A. J., Lugo C. S. B., Elliott T. A., Ware D., Peterson T., Jiang N.✉, Hirsch C. N.✉ and Hufford M. B.✉ (2019). Benchmarking Transposable Element Annotation Methods for Creation of a Streamlined, Comprehensive Pipeline. Genome Biol. 20(1): 275.
Please cite the panEDTA paper if you are using the pan-genome functionality:
Ou S., Collins T., Qiu Y., Seetharam A., Menard C., Manchanda N., Gent J., Schatz M., Anderson S., Hufford M.✉, Hirsch C.✉ (2022). Differences in activity and stability drive transposable element variation in tropical and temperate maize. bioRxiv
Please also cite the software packages that were used in EDTA, listed in the EDTA/bin directory.
You may download the rice genome here (the "all.con" file).
You may want to check out this Q&A page for best practices and get answered. If you have other issues with installation and usage, please check if similar issues have been reported in Issues or open a new issue. If you are (looking for) happy users, please read or write successful cases here.
I want to thank Jacques Dainat for contribution of the EDTA conda recipe as well as improving the codes. I also want to thank Qiushi Li, Zhigui Bao, Philipp Bayer, Nick Carleson, @aderzelle, Sanzhen Liu, Zhougeng Xu, Shun Wang, Nancy Manchanda, Eric Burgueño, Sergei Ryazansky, and many more others for testing, debugging, and improving the EDTA pipeline.




















































































































