jtamames / squeezemeta Goto Github PK
View Code? Open in Web Editor NEWA complete pipeline for metagenomic analysis
License: GNU General Public License v3.0
A complete pipeline for metagenomic analysis
License: GNU General Public License v3.0












































































































Dear SqueezeMeta developers,
Is there any easy way to install SqueezeMeta without sudo privileges, e.g. on a cluster ?
Thanks
Hello,
I was wondering whether there is an option to specify an output directory where all the files SqueezeMeta would output are written? I'm asking this question because I am trying to run SqueezeMeta on an HPC cluster, and I am not allowed to write files where I execute the command.
Best regards,
Sam
Dear Javier,
Thanks a ton for developping SqueezeMeta.
I am trying to use SqueezeMeta in order to generate gene catalog from waste water metagenomes.
I followed the installation instructions but when I run the pipeline on a toy dataset I got the following error :
(squeezemeta) constancias@scelse:/datadrive05/Flo/EZ/NEW_assembly/tests_gene_catalog$ perl /datadrive05/Flo/tools/SqueezeMeta/scripts/SqueezeMeta.pl -m coassembly -p EZ-squeezemeta -s EZ_test_sqeezeMeta.samples -f sub --nobins --nomaxbin --nometabat -t 10
SqueezeMeta v1.0.0 - (c) J. Tamames, F. Puente-Sánchez CNB-CSIC, Madrid, SPAIN
Please cite: Tamames & Puente-Sanchez, Frontiers in Microbiology 9, 3349 (2019). doi: https://doi.org/10.3389/fmicb.2018.03349
Run started Mon Jan 13 16:47:14 2020 in coassembly mode
Now creating directories
Reading configuration from /datadrive05/Flo/EZ/NEW_assembly/tests_gene_catalog/EZ-squeezemeta/SqueezeMeta_conf.pl
7 samples found: ESMetFM01 ESMetFM47 ESMetFM37 ESMetFM09 ESMetFM17 ESMetFM26 ESMetFM06
Now merging files
[0 seconds]: STEP1 -> RUNNING CO-ASSEMBLY: 01.run_assembly.pl (megahit)
Running assembly with megahit
Renaming contigs
Counting length of contigs
Contigs stored in /datadrive05/Flo/EZ/NEW_assembly/tests_gene_catalog/EZ-squeezemeta/results/01.EZ-squeezemeta.fasta
Number of contigs: 86293
[16 minutes, 7 seconds]: STEP2 -> RNA PREDICTION: 02.rnas.pl
Running barrnap (Seeman 2014, Bioinformatics 30, 2068-9) for predicting RNAs: Bacteria[17:03:21] Can't find database: /datadrive04/db/sqeezeMeta/db/test_data/-h/db/bac.hmm
Error running command: /datadrive05/Flo/tools/SqueezeMeta/bin/barrnap --quiet --threads 10 --kingdom bac --reject 0.1 /datadrive05/Flo/EZ/NEW_assembly/tests_gene_catalog/EZ-squeezemeta/intermediate/02.EZ-squeezemeta.maskedrna.fasta --dbdir /datadrive04/db/sqeezeMeta/db/test_data/-h/db > /datadrive05/Flo/EZ/NEW_assembly/tests_gene_catalog/EZ-squeezemeta/temp/bac.gff at /datadrive05/Flo/tools/SqueezeMeta/scripts/02.rnas.pl line 54.
Stopping in STEP2 -> 02.rnas.pl
Died at /datadrive05/Flo/tools/SqueezeMeta/scripts/SqueezeMeta.pl line 663.
Checking the error revealed that I have made a mistake setting up the databases. Actually I wanted to look for help of the download_databases.pl script download_databases.pl -h and it generated another database at the location /datadrive04/db/sqeezeMeta/db/test_data/-h/db.
Then I removed this folder and tried to set up the databases again.
I got these errors
perl /datadrive05/Flo/tools/SqueezeMeta/scripts/preparing_databases/download_databases.pl /datadrive04/db/sqeezeMeta/db/
rm: cannot remove '/datadrive04/db/sqeezeMeta/db/test.tar.gz': No such file or directory
rm: cannot remove '/datadrive05/Flo/tools/SqueezeMeta/lib/classifier.tar.gz': No such file or directory
rm: cannot remove '/datadrive04/db/sqeezeMeta/db/SqueezeMetaDB.tar.gz': No such file or directory
Downloading and unpacking test data...
and I got the same error again running because the script is still looking for the database in the privious location (datadrive04/db/sqeezeMeta/db/test_data/-h/db).
(squeezemeta) constancias@scelse:/datadrive05/Flo/EZ/NEW_assembly/tests_gene_catalog$ perl /datadrive05/Flo/tools/SqueezeMeta/scripts/SqueezeMeta.pl -m coassembly -p EZ-squeezemeta -s EZ_test_sqeezeMeta.samples -f sub --nobins --nomaxbin --nometabat -t 10
Thanks in advance for your help.
Hello all,
I am facing another issue-
/SqueezeMeta/scripts/SqueezeMeta.pl -m Sequential -p test -s test.samples -f mydir --minion
Can't locate Tie/IxHash.pm in @inc (you may need to install the Tie::IxHash module) (@inc contains: /home/mim/miniconda2/lib/site_perl/5.26.2/x86_64-linux-thread-multi /home/mim/miniconda2/lib/site_perl/5.26.2 /home/mim/miniconda2/lib/5.26.2/x86_64-linux-thread-multi /home/mim/miniconda2/lib/5.26.2 .) at /SqueezeMeta/scripts/SqueezeMeta.pl line 12.
BEGIN failed--compilation aborted at /SqueezeMeta/scripts/SqueezeMeta.pl line 12.
Since the "noassembly" flag is supposed to be used with RNA data, such samples should NOT be used for binning, as contigs from the same original genome do not co-vary across samples in metatranscriptomics.
Good morning.
I am running the provided Hadza dataset to test the program but I keep getting the same error.
I installed the databases using the script make_databases.pl and configured them using configure_nodb.pl.
When I run SqueezeMeta using the following:
$LUSTRE/Adri/SqueezeMeta/scripts/SqueezeMeta.pl -m coassembly -p test -s $LUSTRE/Adri/SqueezeMeta/db/test/test.samples -f $LUSTRE/Adri/SqueezeMeta/db/test/raw/ --nopfam -miniden 60
I get this error message:
restart.pl test
[0 seconds]: STEP1 -> RUNNING CO-ASSEMBLY: 01.run_assembly.pl (megahit)
Running assembly with megahit: /mnt/lustre/scratch//home/otras/ini/asm/Adri/SqueezeMeta/scripts/../bin/megahit/megahit -1 /mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/raw_fastq/par1.fastq.gz -2 /mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/raw_fastq/par2.fastq.gz -t 4 -o /mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/megahit
125.0Gb memory in total.
Using: 113.082Gb.
MEGAHIT v1.1.3
--- [Tue Nov 5 12:34:07 2019] Start assembly. Number of CPU threads 4 ---
--- [Tue Nov 5 12:34:07 2019] Available memory: 134912237568, used: 121421013811
--- [Tue Nov 5 12:34:07 2019] Converting reads to binaries ---
[read_lib_functions-inl.h : 209] Lib 0 (/mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/raw_fastq/par1.fastq.gz,/mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/raw_fastq/par2.fastq.gz): pe, 39159256 reads, 100 max length
[utils.h : 126] Real: 49.6343 user: 29.0630 sys: 4.3954 maxrss: 154152
--- [Tue Nov 5 12:34:57 2019] k-max reset to: 119 ---
--- [Tue Nov 5 12:34:57 2019] k list: 21,29,39,59,79,99,119 ---
--- [Tue Nov 5 12:34:57 2019] Extracting solid (k+1)-mers for k = 21 ---
Error running command: /mnt/lustre/scratch//home/otras/ini/asm/Adri/SqueezeMeta/scripts/../bin/megahit/megahit -1 /mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/raw_fastq/par1.fastq.gz -2 /mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/raw_fastq/par2.fastq.gz -t 4 -o /mnt/lustre/scratch/home/otras/ini/asm/Adri/Scripts/Squeeze/test/data/megahit at /mnt/lustre/scratch//home/otras/ini/asm/Adri/SqueezeMeta/scripts/../scripts/01.run_assembly.pl line 83.
Stopping in STEP1 -> 01.run_assembly.pl (megahit)
Any advice will be welcomed.
Thank you,
Adrián.
Hello, all
I am having an error on step 18
[19 hours, 42 minutes, 13 seconds]: STEP18 -> CHECKING BINS: 18.checkM_batch.pl
Creating /home/biofilm/hank/SqueezeMeta/biofilm1/temp/checkm_batch
Reading /home/biofilm/sw/SqueezeMeta/scripts/../data/alltaxlist.txt
Looking for DASTool bins in /home/biofilm/hank/SqueezeMeta/biofilm1/results/DAS/biofilm1_DASTool_bins
39 bins found
Bin 1/39: maxbin.008.fasta.contigs.fa.tax
Using profile for domain rank : Bacteria
Error running command: export PATH="/home/biofilm/sw/SqueezeMeta/scripts/../bin/pplacer":$PATH; /home/biofilm/sw/SqueezeMeta/scripts/../bin/checkm taxon_set domain Bacteria /home/biofilm/hank/SqueezeMeta/biofilm1/data/checkm_markers/Bacteria.ms > /dev/null 2>&1 at /home/biofilm/sw/SqueezeMeta/scripts/../scripts/18.checkM_batch.pl line 136.
Stopping in STEP18 -> 18.checkM_batch.pl
How can I fix the error?
Thanks
Hank
I've got an error I can't quite figure out with checkM:
Bin 132/356: metabat2.338.fa_sub.contigs.fa.tax Using profile for genus rank : Trachipleistophora Using profile for family rank : Pleistophoridae Using profile for rank : Pansporoblastina Error running command: export PATH="/home/parfreylab/Desktop/lab_member_files/programs/SqueezeMeta/scripts/../bin/pplacer":$PATH; /home/parfreylab/Desktop/lab_member_files/programs/SqueezeMeta/scripts/../bin/checkm taxon_set Pansporoblastina /home/parfreylab/Desktop/lab_member_files/morien/bat_guano_metagenomics/bat_guano/data/checkm_markers/Pansporoblastina.ms > /dev/null 2>&1 at /home/parfreylab/Desktop/lab_member_files/programs/SqueezeMeta/scripts/18.checkM_batch.pl line 136.
There is an empty space in the command where the rank should be.
Here's the error if I run that command myself:
checkm taxon_set: error: argument rank: invalid choice: 'Pansporoblastina' (choose from 'life', 'domain', 'phylum', 'class', 'order', 'family', 'genus', 'species')
But, in fact, there is no such input file as Pansporoblastina.ms, so this may be the real problem?
Thanks in advance for your help, devs.
I got the following error at step 6-
Can't locate DBI.pm in @inc.(@inc contains: /home/mim/miniconda2/lib/site_perl/5.26.2/x86_64-linux-thread-multi)
Then I installed DBI.pm in the INC folder linux x86....
Now it gives an error-
Can't locate loadable object for module DBI in @inc (@inc contains: /home/mim/miniconda2/lib/site_perl/5.26.2/x86_64-linux-thread-multi /home/mim/miniconda2/lib/site_perl/5.26.2 /home/mim/miniconda2/lib/5.26.2/x86_64-linux-thread-multi /home/mim/miniconda2/lib/5.26.2 .) at /home/mim/miniconda2/lib/site_perl/5.26.2/x86_64-linux-thread-multi/DBI.pm line 284.
BEGIN failed--compilation aborted at /home/mim/miniconda2/lib/site_perl/5.26.2/x86_64-linux-thread-multi/DBI.pm line 284.
Compilation failed in require at /SqueezeMeta/scripts/../scripts/06.lca.pl line 10.
BEGIN failed--compilation aborted at /SqueezeMeta/scripts/../scripts/06.lca.pl line 10.
Stopping in STEP6 -> 06.lca.pl
KIndly help.
I am getting similar error for /SqueezeMeta/utils/sqm_reads.pl -p gut -s test.samples -f raw/ command also.
Kindly help
As described, this has caused issues for some users.
We should convert the samples file inplace prior to opening it, so that it has *nix line endings. Here are several options:
Apparently it can be done with perl, so we don't need to install dos2unix,
perl -pi -e 's/\r\n/\n/' input.txt
Hello,
When I tested with test data, Squeeze meta was aborted in STEP6. (I used 0.4.2 version however same abort in 0.4.1 version)
[5 hours, 54 minutes, 32 seconds]: STEP6 -> TAXONOMIC ASSIGNMENT: 06.lca.pl
Tax assignment done in /Tools/SqueezeMeta/0.4.2/SqueezeMeta/test_run/results/06.test_run.fun3.tax but no results found. Aborting
Stopping in STEP6 -> 06.lca.pl
How can I pass the STEP6?
Tahnks,
Hello,
When running
module load SqueezeMeta/0.4.3-foss-2018b-Python-2.7.15
cd /user/scratch/gent/gvo000/gvo00043/vsc42339
SqueezeMeta.pl -m coassembly -p squeezemeta_pilot_coassembly_megahit -s /user/scratch/gent/gvo000/gvo00043/vsc42339/pilot.samples -f /user/scratch/gent/gvo000/gvo00043/vsc42339/CLEAN_READS -t 36
I get the following error message:
Cannot find read files in /kyukon/scratch/gent/vo/000/gvo00043/vsc42339/squeezemeta_pilot_coassembly_megahit/data/raw_fastq
Stopping in STEP1 -> 01.run_assembly.pl (megahit)
When I check .../data/raw_fastq however, the paired end read files were copied there by squeezemeta, but they were all 1 KB in size. Then there is a par2.fastq file that is 94 GB in size. I tried to solve this by copying the raw read files to .../data/raw_fastq myself, and running the command again, but the problem seems to persist.
I wonder whether I did something wrong when creating my pilot.samples file? I attached it below. For example, I have two paired end read files for sample PA_4, and I labelled the forward reads as pair1 in the third column, and the reverse reads as pair2 in the third column. So, in total I have 6 samples, each represented by two fastq files, one for the forward reads, and one for the reverse reads. Is this the correct way of doing it?
PA_4 ERR_PA_4_2018_1.fastq pair1
PA_4 ERR_PA_4_2018_2.fastq pair2
PA_5 ERR_PA_5_2018_1.fastq pair1
PA_5 ERR_PA_5_2018_2.fastq pair2
PB_N_7 ERR_PB_N_7_2018_1.fastq pair1
PB_N_7 ERR_PB_N_7_2018_2.fastq pair2
PB_N_8 ERR_PB_N_8_2018_1.fastq pair1
PB_N_8 ERR_PB_N_8_2018_2.fastq pair2
PB_N_9 ERR_PB_N_9_2018_1.fastq pair1
PB_N_9 ERR_PB_N_9_2018_2.fastq pair2
PB_N_OTC_9_10_CON_B ERR_PB_N_OTC9_10_2018_CON_B_1.fastq pair1
PB_N_OTC_9_10_CON_B ERR_PB_N_OTC9_10_2018_CON_B_2.fastq pair2
Or could this be related to the version of SqueezeMeta and the included version of megahit I'm using? I requested the installation of the latest version of SqueezeMeta on the HPC cluster, but since that will take at least a few weeks, or even months, I would like to continue with the version of squeezemeta I currently have at my disposal (v0.4.3)
-p <PROJECT> (as done in v 1.0) but right now we are just reading from ARGV[0]futureDear @fpusan , dear @jtamames ,
I have one question regarrding assembly/binning strategy for metagenomes when co-assembly is not possible/advised.
If I undserstand correctly, in the merged mode of SqueezeMeta, single-assembly is performed and contigs are merged together before mapping and binning.
Could it be possible to use within SqeezeMeta single assembly, mapping of all samples against each of the co-assembly in order to get coverage information which will be used for binning and then per sample binning before checking and collapsing redundancy between MAGs idividualy identified within each sample. This will be similar to what Tom O Delmont et al developped for Tara (see : http://merenlab.org/data/tara-oceans-mags/).
What do you think about this strategy? Would it be feasable to implment it in SqeezeMeta?
Have a great weekend.
Hello,
I have been running squeezemeta for the second time with a slight change in original metatranscriptome fastq samples. I have used coassembly in both cases and run on our local server. However, this time the process is constantly freezing on step 10 and always on Sample#2 (4 PE samples overall), giving this output. The process disappears completely from the server but not giving any error or displaying any change for a long time. The manual interruption causes the process to "wake up" and appear again with the next step ( Aligning to reference with bowtie of Sample2) but being "core dumped" few seconds after and completely exiting. I have tried to repeat the steps using the restart script but did not help, the process freezes again. As the previous run with almost the same samples successfully managed to pass step 10, I am not sure what I could have done wrong. The server seems to have plenty of memory available.
Would be very grateful to hear your advice,
Thank you
[3 days, 1 hours, 40 minutes, 52 seconds]: STEP10 -> MAPPING READS: 10.mapsamples.pl
Reading mapping file from /home/mbogorad/2018/metatrans/unzipped/trimmomatic_analysed/paired/2018metatrans/data/00.2018metatrans.samples
Metagenomes found: 8
Creating reference.
Working with 1: Sample1
Getting raw reads
Aligning to reference with bowtie
Calculating contig coverage
rm: cannot remove '/home/mbogorad/2018/metatrans/unzipped/trimmomatic_analysed/paired/2018metatrans/temp/2018metatrans.Sample1.current_2': No such file or directory
Counting with sqm_counter
37000000 reads counted
Working with 2: Sample2
Getting raw reads
Hi,
When running the Test script, I got this error
Exception in thread "main" java.lang.NoClassDefFoundError: org/apache/commons/io/output/NullWriter
at edu.msu.cme.rdp.classifier.cli.ClassifierMain.main(ClassifierMain.java:67)
Caused by: java.lang.ClassNotFoundException: org.apache.commons.io.output.NullWriter
at java.net.URLClassLoader.findClass(URLClassLoader.java:381)
at java.lang.ClassLoader.loadClass(ClassLoader.java:424)
at sun.misc.Launcher$AppClassLoader.loadClass(Launcher.java:349)
at java.lang.ClassLoader.loadClass(ClassLoader.java:357)
... 1 more
Error running command: java -jar /home/michoug/SqueezeMeta/scripts/../bin/classifier.jar classify /ibex/scratch/michoug/Test/Hadza/temp/16S.fasta -o /ibex/scratch/michoug/Test/Hadza/temp/16S.out -f filterbyconf at /home/michoug/SqueezeMeta/scripts/../scripts/02.run_barrnap.pl line 119.
Stopping in STEP2 -> 02.run_barrnap.pl
Any ideas ?
Dear,
Does squeezeM support data generated from 16S rDNA amplicon sequencing or short gun based NGS data ?
Hello,
At step 14 following error message occured:
Error running command: perl /media/smrtanalysis/Linux_Data/SqueezeMeta/scripts/../bin/MaxBin/run_MaxBin.pl -thread 11 -contig /media/smrtanalysis/Linux_Data/SqueezeMeta/databases/test/ampli_seq01/temp/bincontigs.fasta -abund_list /media/smrtanalysis/Linux_Data/SqueezeMeta/databases/test/ampli_seq01/results/maxbin/abund.list -out /media/smrtanalysis/Linux_Data/SqueezeMeta/databases/test/ampli_seq01/results/maxbin/maxbin -markerpath /media/smrtanalysis/Linux_Data/SqueezeMeta/databases/db/marker.hmm at /media/smrtanalysis/Linux_Data/SqueezeMeta/scripts/../scripts/14.bin_maxbin.pl line 103.
I looked in the code and it stands:
my $command="perl $maxbin_soft -thread $numthreads -contig $tempfasta -abund_list $abundlist -out $dirbin/maxbin -markerpath $databasepath/marker.hmm";
print "Now running Maxbin: $command\n";
my $ecode = system $command;
if($ecode!=0) { die "Error running command: $command"; }
Can you help me out with this error?
Hello!
I'm working with gut metagenomics data and I would like to know...
How should the computer/server be? (for example: RAM, minimum CPU cores, free hard disk, etc.)
It is because I also know that programs like Centrifuge or Kraken uses a lot of RAM memory.
Thanks!!
PD: Could I use CentOs??
Dear SqueezeMeta developers and users,
I have sucessfully run SqeezeMeta to generate a gene catalog and now binning is running on.
I am interested in N cycle pathway and would ultimately like to quantify proportion/abundance of the different genes involved according to my samples.
Thanks
Hi,
Can we do the functional annotation for the Bins extracted from other tools? for example prediction of kegg and metacyc patwhays for each bin ?
Thanks
See #57
Mapping files may contain DOS line endings, which will make it into SQM outputs.
E.g. the 13.*.orftable file in #57 contained a carriage return after a CAZy annotation and before the abundances, and was read as a shorter line by a downstream python script. Hilarity ensues.
We should remove DOS line endings when parsing mapping files, ideally without modifying the original file..
Hello,
I am having an error on step6 LCA
[0 seconds]: STEP6 -> TAXONOMIC ASSIGNMENT: 06.lca.pl
DBD::SQLite::db prepare failed: disk I/O error at /home//metagenomics/2_Scripts/SqueezeMeta/scripts/../scripts/06.lca.pl line 157, line 26.
DBD::SQLite::db prepare failed: disk I/O error at /home//metagenomics/2_Scripts/SqueezeMeta/scripts/../scripts/06.lca.pl line 157, line 26.
Any thing wrong ?
Thanks,
Mohamed
/SqueezeMeta/scripts/SqueezeMeta.pl -m Sequential -s test.samples -f raw/ --minion
SqueezeMeta v1.0.0 - (c) J. Tamames, F. Puente-Sánchez CNB-CSIC, Madrid, SPAIN
Please cite: Tamames & Puente-Sanchez, Frontiers in Microbiology 10.3389 (2019). doi: https://doi.org/10.3389/fmicb.2018.03349
Run started Wed Oct 30 11:26:47 2019 in Sequential mode
Now reading samples
1 metagenomes found
Working with gut
Run started Wed Oct 30 11:26:47 2019 in SEQUENTIAL mode
Now creating directories
Reading configuration from /SqueezeMeta/databases/test/abc/SqueezeMeta_conf.pl
Now linking read files
Now preparing files
ln -s /SqueezeMeta/databases/test/abc/data/raw_fastq/abc.fastq.gz /SqueezeMeta/databases/test/abc/data/raw_fastq/par1.fastq.gz
[0 seconds]: STEP2 -> RNA PREDICTION: 02.run_barrnap.pl
cp: cannot stat '/SqueezeMeta/databases/test/abc/results/01.abc.fasta': No such file or directory
Running barrnap for Bacteria
[11:26:47] Usage: barrnap <file.fasta>
Error running command: /SqueezeMeta/scripts/../bin/barrnap --quiet --threads 12 --kingdom bac --reject 0.1 /SqueezeMeta/databases/test/abc/intermediate/02.abc.maskedrna.fasta --dbdir /SqueezeMeta/databases > /SqueezeMeta/databases/test/abc/temp/bac.gff at /SqueezeMeta/scripts/../scripts/02.run_barrnap.pl line 44.
Stopping in STEP2 -> 02.run_barrnap.pl
I would like to understand two things-
Command for assembly with Canu as it is minion and commands for further analysis.
Kindly help
I'm working on a system that didn't have doMC installed (my oversight, but I'm not admin), so the first time I encountered it, I had the admins install doMC, but the error persists. I've logged out and logged back in to restart the job, and the error is the same. We added a R.Version() command to the dastool R script, and sunk it to a tmp file to check once the error pops up. When I check that file, the R version is 3.5 (the default on the system). When I start R (default, 3.5) in the terminal and load the libarary(doMC), it loads just fine. Devs, any suggestions on what to check next?
Below is my command, edited for brevity:
perl /Linux/SqueezeMeta/scripts/16.dastool.pl projectname
Creating tables of contigs in bins... done
Running DAS Tool for maxbin,metabat2
LD_LIBRARY_PATH=/Linux/SqueezeMeta/scripts/../lib PATH=/Linux/SqueezeMeta/scripts/../bin:$PATH /Linux/SqueezeMeta/scripts/../bin/DAS_Tool/DAS_Tool -i projectname/results/DAS/maxbin.table,projectname/results/DAS/metabat2.table -l maxbin,metabat2 -c projectname/results/01.projectname.fasta --write_bins 1 --score_threshold 0.25 --search_engine diamond -t 32 -o projectname/results/DAS/projectname --db_directory /Archive/Cluster/SqueezeMeta/db
Running DAS Tool using 32 threads.
predicting genes using Prodigal V2.6.3: February, 2016
identifying single copy genes using diamond version 0.9.22
**Error in library(x, character.only = TRUE) :
there is no package called ‘doMC’
Calls: lapply ... suppressPackageStartupMessages -> withCallingHandlers -> library
Execution halted**
Here is the relevant output of the R.Version command we added to the DAStool R script:
$version.string
[1] "R version 3.5.1 (2018-07-02)"
First of all, thank you for the tool. It's making my work easier.
I have a doubt. My samples have triplicates and I inform the SqueezeM in the metadata file what's samples is. Which method does SqueezeM use to merge the triplicates results? Because they give us only one result of raw counts, RPKM, and coverage. Is it an average?
I read your paper about the tool and the item 2 of methods, showing a database creation. How can I access this method?
Thank you very much
I'm finishing up my first test run in SqueezeMeta on a remote cluster and am looking to benchmark it against results from other tools in R on my local machine. I tried to install the SQMtools package via BiocManager, but it says it is unavailable in R version 3.5.2. In the manual it only shows loading the library and I can't find another way to install it locally.
Can you tell me which version it's compatible with, or perhaps another issue that might be happening? The functionality in the vignette looks wonderful so I would love to test it out!
I'm an error when SqueezeMeta try to use the classifier.jar. It say that the jarfile is corrupt.
Running prinseq: /home/grupos/gen/SqueezeMeta/scripts/../bin/prinseq-lite.pl -fasta /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/data/megahit/final.contigs.fa -min_len 200 -out_good /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/results/prinseq; mv /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/results/prinseq.fasta /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/results/01.C-6710.fasta
Input and filter stats:
Input sequences: 149,582
Input bases: 93,788,907
Input mean length: 627.01
Good sequences: 149,582 (100.00%)
Good bases: 93,788,907
Good mean length: 627.01
Bad sequences: 0 (0.00%)
Sequences filtered by specified parameters:
none
Counting length of contigs
Contigs stored in /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/results/01.C-6710.fasta
[26 minutes, 8 seconds]: STEP2 -> RNA PREDICTION: 02.run_barrnap.pl
Running barrnap for Bacteria
Running barrnap for Archaea
Running barrnap for Eukaryote
Running barrnap for Mitochondrial
Running RDP classifier: java -jar /home/grupos/gen/SqueezeMeta/scripts/../bin/classifier.jar classify /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/temp/16S.fasta -o /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/temp/16S.out -f filterbyconf
Error: Invalid or corrupt jarfile /home/grupos/gen/SqueezeMeta/scripts/../bin/classifier.jar
Error running command: java -jar /home/grupos/gen/SqueezeMeta/scripts/../bin/classifier.jar classify /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/temp/16S.fasta -o /SCRATCH/cricasre/Metagenomic/Squeeze/C-6710/temp/16S.out -f filterbyconf at /home/grupos/gen/SqueezeMeta/scripts/../scripts/02.run_barrnap.pl line 120.
Stopping in STEP2 -> 02.run_barrnap.pl
Hi Javier,
It that possible you can upgrade mummer3 to mummer4 in SqueezeMeta?
The nucmer3 uses only one thread and take loooooooong time for large merged assemblies after clustering; nucmer4 use multi-threading without limitation on the size of reference and query sequences.
Or if you can provide guidance to hack your modified minimus2 script to use nucmer4 instead?
Thanks.
Qinglong
Hello,
I was recently running the pipeline and got this error:
sh: 1: cannot create /01.EPS_1.stats: Permission denied
Error running command: /mnt/nfs/bioinfdata/home/NIOO/ohanac/squeezem/SqueezeMeta/scripts/../bin/prinseq-lite.pl -fasta /mnt/nfs/bioinfdata/home/NIOO/ohanac/squeezem/dbs/sPS_1/results/01.EPS_1.fasta -stats_len -stats_info -stats_assembly > /01.EPS_1.stats at /mnt/nfs/bioinfdata/home/NIOO/ohanac/squeezem/SqueezeMeta/scripts/../scripts/01.run_ably.pl line 98.
Stopping in STEP1 -> 01.run_assembly.pl (megahit)
I did a git pull to run the pipeline again in my samples (sequential mode), since I`d like to use the
script sqm2tables.py and summarize my results, but I had this error.
Thanks,
Ohana
Hello,
I have a query regarding use of SqueezeMeta. I have done my analysis till binning using metabat from outside the pipeline SqueezeMeta. Now I want to continue with the pipeline in the mid(after metabat). But I am getting an error "Can't find SqueezeMeta_conf.pl in BjSa_G210. Is the project path ok? at /data/bin/SqueezeMeta-v1.0.0/SqueezeMeta/scripts/16.dastool.pl line 15.
srun: error: slurm-n5: task 0: Exited with exit code 2"
If anyone can help me out to sort this problem.
I am new to metagenome analysis so I need your help if you can.
Thank you,
Hello,
I am having a problem right in the beginning of the pipeline: it seems it its not merging the raw files, even though I can see the links in the raw_fastq folder. I already tried changing the names of the raw files to make them simpler, and I run it with the test data and worked normally.
SqueezeMeta v0.4.2, Jan 2019
Working for sample Control
rm: cannot remove 'home/squeezem/dbs/sip/SIP3/temp/parfast': No such file or directory
Now merging files
cat >
sh: 1: Syntax error: end of file unexpected
rm: cannot remove 'home/squeezem/dbs/sip/SIP3/data/megahit': No such file or directory
Running Megahit for Control: ../../SqueezeMeta/scripts/../bin/megahit/megahit -r --k-list 29,39,59,79,99,119,141 -t 12 -o home/squeezem/dbs/sip/SIP3/data/megahit
1007.0Gb memory in total.
Using: 907.006Gb.
megahit: Cannot find file --k-list
Error running command: ../../SqueezeMeta/scripts/../bin/megahit/megahit -r --k-list 29,39,59,79,99,119,141 -t 12 -o home/squeezem/dbs/sip/SIP3/data/megahit at ../../SqueezeMeta/scripts/../scripts/01.run_assembly_merged.pl line 101.
Stopping in STEP1 -> 01.run_assembly_merged.pl (megahit)
can you help me, please?
thanks
Good evening. I'm running a test with the data provided but having some issues with step 16.
I'm using the restart.pl script
module load cesga/2018 gcccore/6.4.0 gcc/6.4.0 perl/5.28.1 jdk/8u181 ruby/2.6.3 $LUSTRE/Adri/Squeeze2/SqueezeMeta/scripts/restart.pl testing48
but keep getting this error
[0 seconds]: STEP16 -> DAS_TOOL MERGING: 16.dastool.pl Execution halted Can't open /mnt/lustre/scratch/home/otras/ini/asm/Adri/Squeeze2/SqueezeMeta/databases/test/testing48/results/DAS/testing48_DASTool_bins directory
I had already installed the R packages needed (ggplot2, data.table, doMC and DASTool) but testing48_DASTool_bins won't appear in the directory.
The DASTool.log shows the following
predicting genes using Prodigal V2.6.3: February, 2016 identifying single copy genes using diamond version 0.9.22
Thank you very much for your time.
Hello,
I have tried the co-assembly method.
I was looking into the classification details on the files 10.* and 06.*, I noticed that I got the species but not the strain information in my classification. Is-it normal ? and how i can modify to get more information about the strain ?
For instance:
I am expecting to get Rothia mucilaginosa DY-18 in my samples.
I am getting only the information superkingdom:Bacteria;no rank:Terrabacteria group;phylum:Actinobacteria;class:Actinobacteria;order:Micrococcales;family:Micrococcaceae;genus:Rothia;species:Rothia mucilaginosa
I have checked on the alltaxlist.txt file from the tool and the strain is there:
680646 Rothia mucilaginosa DY-18 no rank
Thanks,
Mohamed
Hi,
I'm running SqueezeMeta in co-assembly mode and MaxBin has been using 100% of 1 CPU for about a week, despite -threads being set to 24.
My data are quite large: around 15 samples and 175GB compressed of non-human reads (high sequencing depth). Co-assembly and mapping took around a week to complete.
There may be nothing I can do but wait, but I was wondering is there anything I can do to speed up the process or see how far binning has gotten. Also, is this a common problem with big metagenomic datasets?
Thanks
Hello
I am getting the error on step 15
-bash-4.2$ perl /home//metagenomics/2_Scripts/SqueezeMeta/scripts/restart.pl SqCoa [0 seconds]: STEP15 -> DAS_TOOL MERGING: 15.dastool.pl mkdir: cannot create directory ‘/home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/DAS’: File exists which: no usearch in (/home//metagenomics/2_Scripts/SqueezeMeta/scripts/../bin:/usr/lib64/qt-3.3/bin:/usr/lib64/mpich/bin:/usr/local/cuda/bin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home//.local/bin:/home//bin:/usr/lib64/qt-3.3/bin:/usr/lib64/mpich/bin:/usr/local/cuda/bin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/home//.local/bin:/home//bin:/opt/ncbi-blast-2.7.1+/bin/:::::) /home//metagenomics/2_Scripts/SqueezeMeta/scripts/../bin/DAS_Tool/DAS_Tool: line 241: usearch: command not found mv: cannot stat ‘/home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/03.SqCoa.faa.scg’: No such file or directory mv: cannot stat ‘/home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/03.SqCoa.faa.scg’: No such file or directory rm: cannot remove ‘/home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/03.SqCoa.faa.all.b6’: No such file or directory Error running command: PATH=/home//metagenomics/2_Scripts/SqueezeMeta/scripts/../bin:$PATH /home//metagenomics/2_Scripts/SqueezeMeta/scripts/../bin/DAS_Tool/DAS_Tool -i /home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/DAS/maxbin.table,/home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/DAS/metabat2.table -l maxbin,metabat2 -c /home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/01.SqCoa.fasta --write_bins 1 --proteins /home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/03.SqCoa.faa --score_threshold 0.25 --search_engine diamond -t 38 -o /home//metagenomics/3_Output/metaG/SQUEEZMETA/SqCoa/results/DAS/SqCoa --db_directory /home//metagenomics/1_Input/metaG/Squeezmeta/db at /home//metagenomics/2_Scripts/SqueezeMeta/scripts/../scripts/15.dastool.pl line 63. Stopping in STEP15 -> 15.dastool.pl
In the syslog I have this error:
identifying single copy genes using diamond version 0.9.22
single copy gene prediction using diamond failed. Aborting
identifying single copy genes using diamond version 0.9.22
single copy gene prediction using diamond failed. Aborting
identifying single copy genes using diamond version 0.9.22
single copy gene prediction using diamond failed. Aborting
identifying single copy genes using diamond version 0.9.22
single copy gene prediction using diamond failed. Aborting
Thanks
Hi
Here is the error I got when downloading the main database
Downloading and unpacking database tarball... --2019-07-01 11:31:34-- http://silvani.cnb.csic.es/SqueezeMeta/SqueezeMetaDB.tar.gz Resolving silvani.cnb.csic.es (silvani.cnb.csic.es)... 150.244.82.96 Connecting to silvani.cnb.csic.es (silvani.cnb.csic.es)|150.244.82.96|:80... failed: Connection timed out. Retrying.
Best
Greg
Dear,
Canu is designed to be universal on a large range of PacBio (C2, P4-C2, P5-C3, P6-C4) and Oxford Nanopore (R6 through R9) data. And, SqueezeMeat uses Canu for assembly of the long, error-prone MinION reads. So, does SqueezeMeat support PacBio long reads ?
What would be the best strategy to compare the genes proportion among different samples. can I directly compare tpm values or should I subsample the count values (e.g., KEGG abundance data) to the same number of sequences per sample and compare those 'rarefied' values?
Should I take into consideration the sample's mapping rates against the coassembly?
Hi Fernando and Javier:
I've updated SQM again it works great,except for the fact there is a new error when I call the sqm2tables.py.
The error says this:
File "/home/install/SqueezeMeta-1.0.0/utils//sqm2tables.py", line 174, in
main(parse_args())
File "/home/install/SqueezeMeta-1.0.0/utils//sqm2tables.py", line 63, in main
sampleNames, orfs, kegg, cog, pfam, custom = parse_orf_table(perlVars['$mergedfile'], nokegg, nocog, nopfam, args.trusted_functions, args.ignore_unclassified, customMethods)
ValueError: not enough values to unpack (expected 6, got 5)
Thanks and have a nice day!
Rafa
Hi,
I have an external assembly of several samples that I will concatenate and give to the software with the -extassembly options.
Is the cd-hit step done prior to further analysis or should I do it myself?
Best
Greg
Hello, I'm having this problem at step 6. Both DBI and gcc are installed and I have added them to the $PATH. any ideas to solve this issue?
[0 seconds]: STEP6 -> TAXONOMIC ASSIGNMENT: 06.lca.pl
Can't locate DBI.pm in @inc (you may need to install the DBI module) (@inc contains: /home/leticia/anaconda3/lib/site_perl/5.26.2/x86_64-linux-thread-multi /home/leticia/anaconda3/lib/site_perl/5.26.2 /home/leticia/anaconda3/lib/5.26.2/x86_64-linux-thread-multi /home/leticia/anaconda3/lib/5.26.2 .) at /home/leticia/SqueezeMeta-1.0.0/scripts/../scripts/06.lca.pl line 10.
BEGIN failed--compilation aborted at /home/leticia/SqueezeMeta-1.0.0/scripts/../scripts/06.lca.pl line 10.
Stopping in STEP6 -> 06.lca.pl
Thanks :)
Leticia
Hello!
I have some questions about DIAMOND output and taxonomy assignation with SQM_reads. I searched an specific sequence in .tax.wrank file (already filtered), .tax.m8 DIAMOND output file and in .fastq initial file, for doing blastx manually in NCBI:
[user@fs6803]$ seqid='b41fc6c6-2e98-46ee-ac43-cf114a7aa921'
[user@fs6803]$ grep $seqid SAMP10.fastq.gz.tax.wranks
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 k_Bacteria;n_FCB group;n_Bacteroidetes/Chlorobi group;p_Bacteroidetes;c_Bacteroidia;o_Bacteroidales;f_Rikenellaceae;g_Alistipes;s_Alistipes sp.
[user@fs6803]$ grep $seqid SAMP10.fastq.gz.tax.m8
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 CCY34802.1 40.8 174 83 8 729 250 220 387 9.4e-13 84.0
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_010420406.1 37.5 176 86 8 729 250 220 387 1.5e-10 76.6
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_088655160.1 36.8 174 90 6 729 250 220 387 2.5e-10 75.9
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_010662461.1 38.7 173 88 8 729 250 220 387 4.3e-10 75.1
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_062126048.1 37.2 172 92 8 729 250 220 387 7.4e-10 74.3
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_009216967.1 33.3 180 88 7 729 250 220 387 1.8e-08 69.7
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_106152287.1 36.0 172 94 6 729 250 220 387 3.1e-08 68.9
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 RKV88849.1 33.3 180 88 7 729 250 220 387 5.3e-08 68.2
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 KWW31213.1 34.9 175 92 7 729 250 220 387 9.1e-08 67.4
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_010527157.1 32.9 173 98 8 729 250 220 387 1.5e-07 66.6
b41fc6c6-2e98-46ee-ac43-cf114a7aa921 WP_036826742.1 32.4 170 102 6 729 250 220 386 7.7e-07 64.3
[user@fs6803]$ zcat $PTH/SAMP10.fastq.gz | grep -A1 $seqid
@b41fc6c6-2e98-46ee-ac43-cf114a7aa921 runid=dfc10e4b4b18bdfab43489ccc2ca2f7a3c0fe9f8 sampleid=run37 read=53938 ch=2 start_time=2019-07-17T12:33:59Z barcode=barcode10
CGGGCAGCGGTAACGTCAACCGAACCGTGGAACATGATACCCACTTCGG(...)GGTTATTCGCCGAAACGAC
Here, some of the blastx outputs:
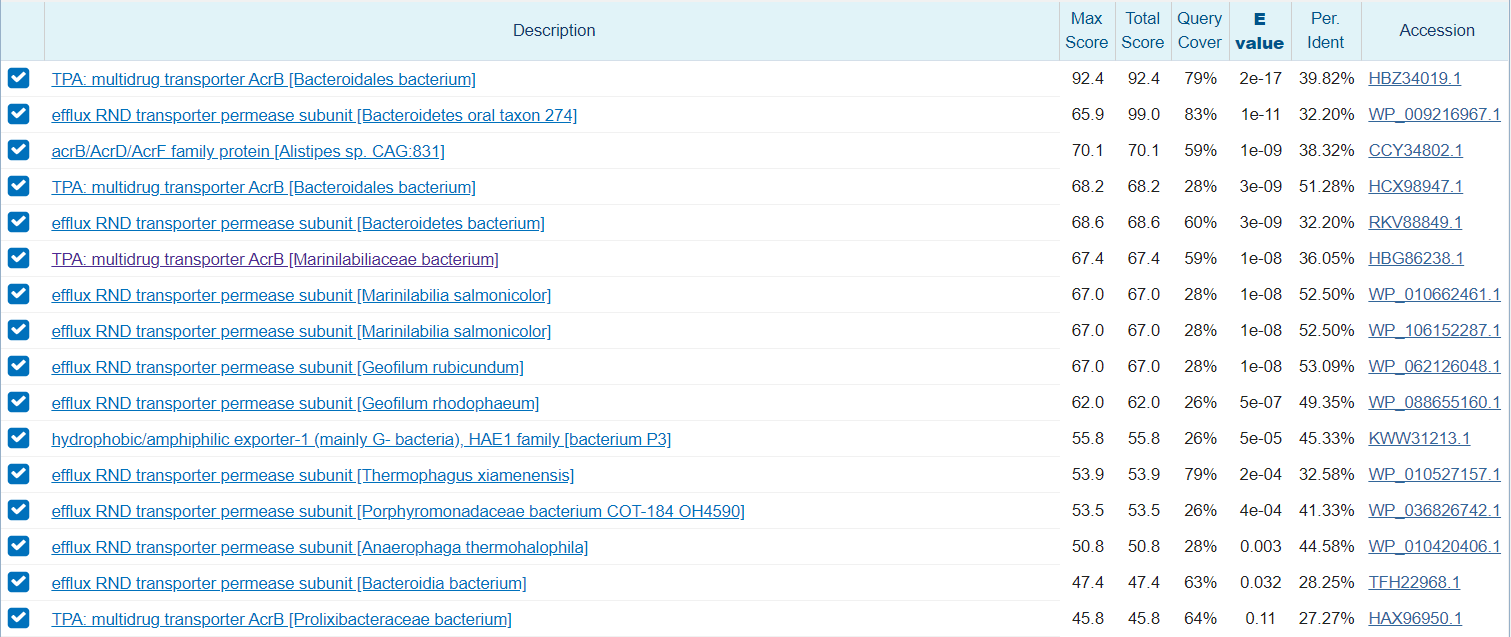
It calls my attention that SQM_reads is classifying this sequence as "s_Alistipes sp" with a maximum identity percentage of 40.8 % (if I am correctly interpreting the equivalence between blastx and SQM_reads DIAMOND outputs). As I understand, with identity filters and posterior LCA algorithm
this sequence should not be assigned to species level.
Am I misinterpreting outputs or is there some kind of error in SQM_reads taxonomy?
Thanks a lot!
Hello,
I am a new user to Squeezemeta and thus I need a basic help. I have installed everything and have all the dependencies.
Eventhough the commands have been well explained, I am facing an issue there.
For e.g- /SqueezeMeta/scripts/01.run_assembly.pl -m Sequential -p test -s test.samples -f mydir –minion
I am getting the following error-
Can't find SqueezeMeta_conf.pl in -m. Is the project path ok? at /SqueezeMeta/scripts/01.run_assembly.pl line 15.
Can someone guide me with the write way of writing commands.
Good day
I have tried running the sqm2itol.pl with the following command:
$ sqm2itol.pl -p sandsamples_10hrs_coassembly/ -completion 50 -contamination 10 -classification kegg -function /home/kinosha/functions_10
However I keep getting this same error and I am not sure what is the problem is and how to fix it.
Reading bins in /home/kinosha/sandsamples_10hrs_coassembly/results/19.sandsamples_10hrs_coassembly.bintable...
Found 78 bins
Working with 34 bins with more than 50 % completion and less than 10 % contamination
Reading requested functions from /home/kinosha/functions_10
Reading functions in /home/kinosha/sandsamples_10hrs_coassembly/results/21.sandsamples_10hrs_coassembly.kegg.pathways
Reading sequences in /home/kinosha/sandsamples_10hrs_coassembly/results/03.sandsamples_10hrs_coassembly.faa
Getting sequences for bin metabat2.98.fa.contigs_
Running CompareM: comparem aai_wf --proteins /home/kinosha/sandsamples_10hrs_coassembly/temp/comparem -x faa /home/kinosha/sandsamples_10hrs_coassembly/results/maxbin
Died at /apps/SqueezeMeta/utils/sqm2itol.pl line 213.
Please could someone give me some assistance.
Thanking you in advance
Hello. I am trying to run a single end whole genome metagenome sample sequenced on nanopore and trying to analyze on squeezemeta.
Can someone guide me on the exact format and correct file format of the test.sample file.
Useful pipeline. Seems to be crashing at line 136 of the checkM script and it appears to be an issue with the CheckM markers (nothing in the checkm_markers folder).
Bin 1/77: maxbin.077.fasta.contigs.fa.tax
Using profile for order rank : Clostridiales
Error running command: export PATH="/home/mdjlynch/bioinformatics-tools/SqueezeMeta/scripts/../bin/pplacer":$PATH; /home/mdjlynch/bioinformatics-tools/SqueezeMeta/scripts/../bin/checkm taxon_set order Clostridiales /home/mdjlynch/working/pilot-1-to-6-ensemble-binning/data/checkm_markers/Clostridiales.ms > /dev/null 2>&1 at /home/mdjlynch/bioinformatics-tools/SqueezeMeta/scripts/../scripts/18.checkM_batch.pl line 136.
Stopping in STEP18 -> 18.checkM_batch.pl
Thoughts?
Thanks.
Hi Javier, I'm having problems with the sqm2tables.py script. I've just upgraded the old script with the one that reads the trnas with aragorn and also change the util.py to the latest one in your git.
It runs well until the end where it shows this error:
File "/home/install/SqueezeMeta-1.0.0/utils//sqm2tables.py", line 164, in
main(parse_args())
File "/home/install/SqueezeMeta-1.0.0/utils//sqm2tables.py", line 128, in main
write_orf_seqs(orfs['abundances'].keys(), perlVars['$aafile'], fna_blastx, perlVars['$rnafile'], perlVars['$trnafile'] + '.fasta', prefix + 'orf.sequences.tsv')
KeyError: '$trnafile'
All the tables are made except for orfs.sequences.tsv, prokfilter.abund.tsv and allfilter.abund.tsv.
Thank you and have a nice day!
Rafa
I have SqueezeMeta installed on CentOS7 with conda. I've been trying to run the test data before using my own dataset, but I am running into a bug where step 18 hangs. I think I've tracked the bug down to checkM data setRoot:
The initial error:
[5 hours, 17 minutes, 40 seconds]: STEP18 -> CHECKING BINS: 18.checkM_batch.pl
Creating /home/ben/test_squeeze/hadza/temp/checkm_batch
Reading /home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/data/alltaxlist.txt
Looking for DASTool bins in /home/ben/test_squeeze/hadza/results/DAS/hadza_DASTool_bins
23 bins found
Bin 1/23: maxbin.045.fasta.contigs.fa.tax
Using profile for domain rank : Bacteria
Error running command: PATH=/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin:/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/pplacer:/home/b
en/.conda/envs/SqueezeMeta/SqueezeMeta/bin/hmmer:$PATH /home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/checkm taxon_set domain Bacteria /home/be
n/test_squeeze/hadza/data/checkm_markers/Bacteria.ms > /dev/null 2>&1 at /home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/scripts/18.checkM_batch.pl
line 143.
Stopping in STEP18 -> 18.checkM_batch.pl
Running /home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/checkm taxon_set domain Bacteria /home/ben/test_squeeze/hadza/data/checkm_markers/Bacteria.ms manually:
It seems that the CheckM data folder has not been set yet or has been removed. Running: 'checkm data setRoot'.
Where should CheckM store it's data?
Please specify a location or type 'abort' to stop trying:
/data/shared_databases/squeezedb
*******************************************************************************
[CheckM - data] Check for database updates. [setRoot]
*******************************************************************************
Path [/data/shared_databases/squeezedb] exists and you have permission to write to this folder.
(re) creating manifest file (please be patient).
Unexpected error: <class 'TypeError'>
Traceback (most recent call last):
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/checkm", line 718, in <module>
checkmParser.parseOptions(args)
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/main.py", line 1229, in parseOptions
self.updateCheckM_DB(options)
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/main.py", line 82, in updateCheckM_DB
DBM.runAction(options.action)
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/checkmData.py", line 122, in runAction
path = self.setRoot(path=action[1])
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/checkmData.py", line 140, in setRoot
path = self.confirmPath(path=path)
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/checkmData.py", line 193, in confirmPath
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/manifestManager.py", line 85, in createManifest
self.walk(parents, root_path, '', dirs, files, skipFile=manifestName)
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/manifestManager.py", line 330, in walk
self.hashfile(path),
File "/home/ben/.conda/envs/SqueezeMeta/SqueezeMeta/bin/../lib/checkm/manifestManager.py", line 364, in hashfile
hasher.update(buf)
TypeError: Unicode-objects must be encoded before hashing
Through trial and error I've found that I can manually set the data root to any empty directory. I only receive a TypeError if there are files inside the directory.
Thanks for all of your hard work developing SqueezeMeta! 😃
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.